Fibrous rings of heart valves in undifferentiated connective tissue dysplasia
Bibliographic description:
Kuznetsova, V.V. Fibrous rings of heart valves in undifferentiated connective tissue dysplasia / V.V. Kuznetsova.
— Text: direct // Medicine and healthcare: materials of the I International. scientific conf. (Chita, November 2012). - Chita: Young Scientist Publishing House, 2012. - pp. 26-36. — URL: https://moluch.ru/conf/med/archive/62/2893/ (access date: 12/27/2021). In the literature there are reports on the diameter of the mitral and aortic fibrous rings in connective tissue dysplasia. According to research by Yakovlev V.M. et al. [5], an increase in the diameter of the mitral annulus fibrosus in both phases of the cardiac cycle can serve as a criterion for connective tissue dysplasia of the mitral valve with impaired function. This work revealed a statistically significant increase in the diameter of the mitral fibrous annulus in both phases of the cardiac cycle in the group of patients with mitral valve prolapse with the presence of mitral regurgitation in comparison with the control group (patients without connective tissue dysplasia) and within the experimental group (patients with mitral valve prolapse without mitral regurgitation). In the work of Maleev E.G et al. [1] Patients with mitral valve prolapse also had a larger mitral annulus diameter compared to controls.
In JR Matos-Souza et al. [6] examined 627 people with isolated mitral valve prolapse and 627 people matched by age, gender and body mass index without mitral valve prolapse; a total of 454 men and 800 women aged 37.9 ± 0.3 with a body mass index of 23.7 ± 0.1 kg/m2. Individuals with mitral valve prolapse had a significantly larger diameter of the aortic annulus compared with controls (30.4 ± 0.1 versus 29.5 ± 0.1 mm). In addition, multivariate analysis demonstrated an independent association between mitral valve prolapse and aortic annulus size in a model that included the confounding variables age, sex, body mass index, body surface area, blood pressure, and left ventricular myocardial mass index [6].
Regarding the diameter of the other two fibrous rings of the heart in connective tissue dysplasia, no data were found in the available literature.
Purpose of the study:
study of the diameter of the fibrous rings of the heart valves in connective tissue dysplasia and their relationship with anthropometric indicators and morpho-functional indicators of the heart.
Material and methods.
The study is based on data obtained from examining 60 patients with CTD aged from 18 to 44 years (average age - 25.9±6.8 years) and 27 patients without signs of CTD. An objective study was carried out according to generally accepted methods. During the physical examination, the following measurements were taken: height, weight, chest circumference (CHC), waist circumference (WC), arm span, sitting height, hand length, foot length. Based on the data obtained, BMI, body surface area (BSA), proportionality indices (PI between OGK and standing height, PI between leg length and torso length, Pigne index) were calculated. The patients' SBP, DBP and heart rate were determined.
The presence of connective tissue dysplasia in patients was determined by a combination of signs [3]. Doppler echocardiography was carried out on an expert-class ultrasound scanner with color Doppler mapping VIVID-3 from General Electric (USA). Ultrasound examination of the heart was performed in accordance with the recommendations of the European and American Associations of Echocardiography for measurements, calculations and evaluation of the chambers of the heart and great vessels (2006). The location was carried out in the parasternal position (III-IV intercostal space along the left edge of the sternum) in a horizontal position of the patient with the head end elevated by changing the angle of the sensor for sequential imaging of various parts of the heart. To obtain the structural characteristics of the heart, we assessed the final diastolic and systolic diameters (LV EDV, LV ESD, cm), volumes (EDV, ESV, ml) of the LV, the thickness of the posterior wall of the LV (LV TZW, cm) in systole and diastole, the thickness of the interventricular septum ( VSD, cm) in systole and diastole, indexed indicators of LV volumes (ICS, ICDO, ml/m2), LV myocardial mass (LVMM, g). Structural and geometric changes were determined by the LVMM index (LVMI, g/m2), the relative thickness of the walls of the left ventricle and myocardial stress. Left ventricular volumetric parameters and left ventricular ejection fraction were calculated using the Teichholz formulas [4]. Indicators of left ventricular systolic function included: ejection fraction (EF, %), fraction of shortening of the anterior-posterior diameter of the LV in systole (FU, %), velocity of circular shortening of myocardial fibers (Vcf, s-1). When assessing the state of central and peripheral hemodynamics, we took into account the values of stroke (SV, ml) and minute (MR, l/min) volumes, stroke (ml/m2) and cardiac (l/min/m2) indices, total peripheral vascular resistance (TPVR, din*s*cm-5). The diastolic function of the left ventricle was studied according to the time and speed parameters of the transmitral diastolic flow: isovolumetric relaxation time (IVR, s), acceleration time of the early transmitral diastolic flow (AT, s), deceleration of the early transmitral diastolic flow (DT, s), ejection period (ET) , s), peak speeds of fast (E, m/s) and slow (A, m/s) diastolic filling, their ratio (E/A) [4].
Statistical data processing was carried out using the Statistica 6.0 application package from StatSoft and MIX for Windows, as well as Microsoft Excel capabilities. Data analysis for normality of distribution was carried out using the Kolmogorov-Smirnov test. The hypothesis of normal distribution of the quantitative trait was accepted as the null hypothesis. Since the distribution of all characteristics was different from the normal distribution, the characteristics were described by the median and interquartile range (25th and 75th percentiles). Comparisons between two independent groups were performed using the Mann-Whitney U test. The relationship between two variables was assessed using the Spearman correlation coefficient. The level of acceptance or rejection of the null hypothesis was below 0.05.
Results and discussion.
General characteristics of the compared groups. Patients with DST (Table 1) had significantly greater body length compared to the control group (p=0.0001) and significantly lower body weight (p=0.0004), which is a reflection of the general properties of the population of patients with connective tissue dysplasia. At the same time, BMI and BSA in patients with CTD were statistically significantly lower than in patients in the control group (p<0.0001 and p=0.02). WC and OGK in patients with DST were statistically significantly lower than in patients in the control group (p=0.00001 and p=0.0005). Sitting height, hand and foot length, and arm span were not statistically significantly different between groups. SBP, DBP and heart rate in patients with CTD were statistically significantly reduced compared to the control group (p=0.007, p=0.03 and p=0.02, respectively).
Table 1
General characteristics of patients in the study groups, Me (P25-P75)
| Index | Patients with DST ( n =149) | Control group ( n=55) | p |
| Height, cm | 176 (170-183) | 170 (161-177) | 0,0001 |
| Sitting height, cm | 92 (90-96) | 93 (90,5-96,5) | 0,5 |
| OGK, cm | 79 (73-86) | 86 (80-94) | 0,0005 |
| OT, cm | 70 (67-76) | 79 (75,5-88) | 0,00001 |
| Arm span, cm | 179 (173-186) | 181 (171-186,5) | 0,8 |
| Brush, cm | 20 (19-21) | 20 (19-21) | 0,77 |
| Foot, cm | 27(26-29) | 28 (25,5-29) | 0,79 |
| Weight, kg | 60 (53-68) | 66 (57-76) | 0,0004 |
| BMI, kg/m2 | 19,3 (17,7-21,2) | 22,8 (21,2-25,6) | <0,00001 |
| PPT, m2 | 1,76 (1,62-1,89) | 1,81 (1,72-1,94) | 0,02 |
| SBP, mmHg | 100 (90-110) | 110 (100-120) | 0,007 |
| DBP, mmHg | 60 (55-70) | 65 (60-80) | 0,03 |
| Heart rate, beats/min | 70 (64-75) | 77 (69-84) | 0,02 |
Thus, patients with DST were anthropometrically significantly different from the control group. Patients with DST were characterized by significantly greater height, lower body weight, BMI, body surface area, OGK, WC, which is typical for the population of these patients as a whole.
When comparing the structural and geometric parameters of the heart in patients with CTD and individuals in the control group, a number of differences were revealed (Table 2).
The dimensions of all heart cavities, measured in diastole, in patients with CTD were significantly smaller than in patients in the control group (p<0.05), which is explained by the smaller body size in patients with CTD. LV EDV and indexed LV EDV in patients with DST were also significantly reduced compared to control values (p=0.008 and p=0.03). Thus, with the same body surface area, patients with DST were characterized by lower LV EDV. At the same time, there was no significant decrease in LV ESR (p = 0.44), ESV (p = 0.32) and LV ICSV (p = 0.89). The thickness of the IVS and LVSD in diastole did not differ significantly between the groups (p = 0.32 and p = 0.19); in systole, the thickness of the IVS and LVSD was significantly less in patients with DST (p = 0.02 and p = 0.002 ). Relative left ventricular wall thickness did not differ between groups (p=0.37). LVMM and LVMI were significantly lower in the group with DST (p=0.03 and p=0.045). The diameter of the inferior vena cava in the group with DST was underestimated compared to that in the control group (p=0.04). There were no significant differences in the magnitude of the CSMR in the compared groups (p=0.21).
table 2
Structural and geometric characteristics of the heart of patients with DST and the control group, Me (P25-P75)
| Index | Patients with DST ( n= 60) | Control group ( n= 27) | p |
| LV EDV, cm | 4,5(4,23-4,82) | 4,8(4,5-5,0) | 0,009 |
| LV ESD, cm | 3,0(2,7-3,19) | 3,1(2,8-3,2) | 0,44 |
| IVS s, cm | 1,17(1,01-1,27) | 1,25(1,15-1,35) | 0,02 |
| IVD d, cm | 0,82(0,7-0,92) | 0,85(0,8-0,91) | 0,32 |
| LVL s, cm | 1,29(1,2-1,4) | 1,4(1,3-1,5) | 0,002 |
| LVL d, cm | 0,82(0,75-0,93) | 0,86(0,8-0,96) | 0,19 |
| Rel. LV wall thickness | 0,35 (0,32 -0,39) | 0,34 (0,33-0,38) | 0,37 |
| LP, cm | 2,9(2,6-3,18) | 3,3(3,0-3,5) | 0,0004 |
| PP, cm | 3,18(2,9-3,46) | 3,4(3,27-3,6) | 0,003 |
| NPV, cm | 1,06(0,81-1,85) | 1,8(1,43-2,0) | 0,04 |
| RV, cm | 2,1(1,9-2,38) | 2,4(2,0-2,63) | 0,02 |
| LVMM, g | 119,7(95,29-146) | 135,5(117,1-151) | 0,03 |
| LV EDV, ml | 92,75 (79-108,5) | 108 (92,84-118) | 0,008 |
| LV ESV, ml | 35 (27,27-39,66) | 38 (30-41) | 0,32 |
| LV ICD, ml/m2 | 53,65(48,88-61,9) | 58,06(52,45-67,05) | 0,03 |
| LV ICS, ml/m2 | 20,31(16,08-22,54) | 20,03(17,2-22,68) | 0,89 |
| LVMI, g/m2 | 72,29(59,67-82,38) | 74,86(69,9-87,68) | 0,045 |
| LV CVMS, dyn*cm2 | 109,56(95,13-123,99) | 112,92(106,02-122,38) | 0,21 |
According to the literature, various indicators of the state of transaortic and transpulmonary blood flow give an idea of the systolic function of the left and right ventricles[2]. At the same time, indicators of transmitral and transtricuspid diastolic blood flow reflect the diastolic function of the left and right ventricles (Table 3).
Peak transaortic velocity and peak transaortic pressure gradient in the group of patients with CTD were statistically significantly lower than in the control group (p=0.005 and p=0.004). Peak transmitral velocity and peak transmitral pressure gradient did not differ between groups (p=0.51 and p=0.39). There were also no statistically significant differences in peak transtricuspid velocity and peak transtricuspid pressure gradient (p=0.97 and p=0.97). Peak transpulmonary velocity and peak transpulmonary pressure gradient in the group with DST were significantly reduced compared to control values (p=0.006 and p=0.007).
Table 3
Transvalvular peak velocity and pressure gradients of patients with DST and the control group, Me (P25-P75)
| Indicators | Patients with DST (n=60) | Control group (n=27) | p |
| Peak transaortic velocity, m/s | 1,07(0,95-1,18) | 1,17(1,03-1,3) | 0,005 |
| Peak transaortic pressure gradient, mm Hg. Art. | 4,5(3,63-5,6) | 5,48(4,24-6,76) | 0,004 |
| Peak transmitral velocity, m/s | 0,79(0,72-0,9) | 0,8(0,76-0,9) | 0,51 |
| Peak transmitral pressure gradient, mm Hg. Art. | 2,51(2,05-3,24) | 2,56(2,31-3,24) | 0,39 |
| Peak transpulmonary velocity, m/s | 0,89(0,77-0,99) | 0,95(0,9-1,16) | 0,006 |
| Peak transpulmonary pressure gradient, mm Hg. Art. | 3,14(2,39-3,92) | 3,61(3,21-5,38) | 0,007 |
| Peak transtricuspid velocity, m/s | 0,71(0,62-0,78) | 0,7(0,63-0,77) | 0,97 |
| Peak transtricuspid pressure gradient, mm Hg. Art. | 1,96(1,55-2,45) | 1,96(1,59-2,37) | 0,97 |
When studying the contractile function of the left ventricle in patients with CTD and the control group, regular differences were also revealed (Table 4).
LVEF and LVEF were reduced in patients with CTD compared to the control group, but the differences were not statistically significant (p=0.18 and p=0.09, respectively). However, a trend towards a decrease in the left ventricular shortening fraction in patients with CTD can be noted. The rate of circular shortening of myocardial fibers in patients with CTD was significantly lower than in the control group (p = 0.0007). The percentage of systolic shortening of the IVS and LVSD did not differ significantly between groups (p=0.30 and p=0.7).
Table 4
Indicators of LV systolic function in patients with DST and the control group, Me (P25-P75)
| Indicators | Patients with DST ( n =60) | Control group ( n =27) | p |
| EF, % | 64,18 (60,28-68,01) | 65,4 (62-72) | 0,18 |
| UGH, % | 34,82 (31,74-37,78) | 36,36(33,33-41,67) | 0,09 |
| Rate of circular shortening of myocardial fibers, s-1 | 0,11 (0,10-0,13) | 0,13 (0,12 -0,15 ) | 0,0007 |
| % systolic shortening of the IVS | 41,33 (29,47-55,56) | 46,34 (30,53-60) | 0,39 |
| % systolic shortening of the IVS | 56,17 (40,91-73,33) | 60 (50,52-66,67) | 0,7 |
The main integrative indicators characterizing the work of the heart are SV and MO of the ventricles. In individuals with DST, the most important parameters of the myocardial pumping function were underestimated compared to control indicators (Table 5).
Table 5
State of central and peripheral hemodynamics in patients with DST and the control group, Me (P25-P75)
| Indicators | Patients with DST ( n =60) | Control group ( n =27) | p |
| UO, ml | 57,2 (52-69,34) | 70 (61-81) | 0,003 |
| MOC, l/min | 4,08 (3,4-4,82) | 5,46 (4,31-6,24) | 0,0001 |
| UI, ml/m2 | 34,62 (30,87-39,27) | 38,1 (34,78-45,25) | 0,01 |
| SI, l/m2*min | 2,38 (2,03-2,9) | 3,03 (2,55-3,33) | 0,0006 |
| OPSS, dyn*s*cm-5 | 1483,15(1275,5-1789,42) | 1224,39(1103,17-1534,85) | 0,01 |
In persons with DST, all indicators characterizing the functioning of the left ventricle were significantly reduced: SV (p = 0.003), MV (p = 0.0001), CI (p = 0.01), CI (p = 0.0006). OPSS in the group with DST significantly exceeded control values (p = 0.01), which can be explained by a compensatory reaction to a decrease in cardiac output.
Thus, the contractile process of the left ventricle in patients in our sample, compared with the control group, was characterized by a decrease in the speed of circular contraction of myocardial fibers, peak transaortic velocity and the pressure gradient that determines it, SV, MOC, CI, CI. Compensatory reactions from peripheral hemodynamics were characterized by an increase in peripheral vascular resistance.
In recent years, the attention of many researchers of cardiac hemodynamics has been focused on diastolic function, changes in which are the earliest in the pathophysiology of cardiac hemodynamic disorders (Table 6).
In patients with CTD, LV VIR (IVRT) was significantly reduced compared to the control group (p=0.01). The acceleration time of early transmitral diastolic flow AT in patients with DST was statistically significantly less than in the control group (p=0.001). The deceleration time of early transmitral diastolic flow DT, which responds to changes in myocardial stiffness, did not differ between groups, although there was a tendency for it to increase in the group of patients with DST (p = 0.07). The total time of early and late diastolic flow turned out to be statistically significantly longer in the group of patients with DST (p=0.002) due to the lengthening of the time of late diastolic flow. Early diastolic flow time was not significantly different between groups (p=0.38). Thus, in patients with DST, the increase in diastole duration occurred due to the atrial systole phase, that is, diastole was more energy-intensive. Differences in velocity indicators of LV diastolic function in individuals with CTD were represented by a decrease in the atrial component of transmitral diastolic flow (p = 0.01), which resulted in an increase in the ratio of velocity flows (p = 0.075). Thus, the patients in our sample were characterized by impaired diastolic filling of the left ventricle, which was caused by accelerated relaxation, an increase in the period of late diastolic filling and a relative shortening of the period of early diastolic filling.
Table 6
Diastolic function of the left ventricle in patients with DST and the control group, Me (P25-P75)
| Indicators | Patients with DST ( n= 60) | Control group ( n= 27) | p |
| IVRT, ms | 72 (64-80) | 80 (73-88) | 0,01 |
| AT, ms | 72 (60-87) | 88 (80-104) | 0,001 |
| DT, ms | 150(57-192) | 111(44-136) | 0,07 |
| Time of early and late diastolic filling, ms | 501 (444-589) | 430 (358-494) | 0,002 |
| Early transmitral diastolic flow time, ms | 225,5(132-284) | 199 (159-236) | 0,38 |
| Early diastolic filling speed, m/s | 0,79(0,72-0,90) | 0,8 (0,76-0,9) | 0,51 |
| Late diastolic filling rate, m/s | 0,48(0,43-0,58) | 0,6(0,47-0,63) | 0,01 |
| E/A | 1,56(1,38-1,89) | 1,4(1,23-1,73) | 0,08 |
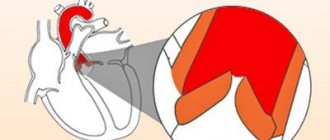
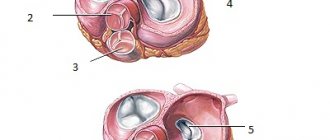
The fibrous skeleton of the heart is formed by four rings interconnected by connective tissue. The atria, ventricles, valves, pulmonary arterial trunk and aorta are tightly attached to this connective tissue framework. In patients with CTD, ROS was significantly less than in patients in the control group (p = 0.002), which can be explained by the smaller size of the body and, accordingly, the heart of patients with CTD (Table 7). At the same time, in patients with DST, a pathological expansion of the IFC was observed, exceeding control values (p = 0.007). According to the data presented in [6], this indicator can serve as a criterion for connective tissue dysplasia of the mitral valve with impairment of its function. Tricuspid and pulmonary annulus fibrosus did not differ between groups (p=0.46 and p=0.8).
Table 7
Diameter of fibrous rings of valves in patients with DST and control group, Me (P25-P75)
| Indicators | Patients with DST ( n =60) | Control group ( n =27) | p |
| AFK, cm | 1,86 (1,73-2,05) | 2,01(1,9-2,3) | 0,002 |
| MFK, cm | 3,57 (3,27-3,81) | 3,2(3,0-3,45) | 0,007 |
| TFC, cm | 2,7 (2,5-3,2) | 2,9(2,8-3,0) | 0,46 |
| PFC, cm | 2,1 (1,9-2,25) | 2,05(1,9-2,25) | 0,8 |
For ROS, positive correlations of moderate strength were obtained with body size (weight (r=0.41, p=0.001), BMI (r=0.38, p=0.003), body surface area (r=0.36, p= 0.005), OGK (r=0.34, p=0.009), OT (r=0.4, p=0.002), proportionality index OGK/growth (r=0.3, p=0.02)), indicators preload (LV EDV (r=0.46, p=0.0003), LV EDC (r=0.41, p=0.001)), central hemodynamic parameters (LV SV (r=0.51, p=0, 00005), IOC (r=0.50, p=0.00007)), LVMM (r=0.43, p=0.0008), indicators of LV systolic function (IVS thickness in systole (r=0.43, p=0.0008), LVAD thickness in systole (r=0.42, p=0.001).Negative correlations of moderate strength were obtained for AFC and OPSS (r=-0.41, p=0.002), Pignier index (r =-0.38, p=0.005 ).
Positive correlations of moderate strength were identified between the diameter of the IFC and body size (height (r=0.31, p=0.02), weight (r=0.33, p=0.02), body surface area (r= 0.37, p=0.007)), left ventricular ejection time (r=0.48, p=0.0004), total time of early and late transmitral diastolic flow (r=0.34, p=0.01) , negative correlations of average strength between the diameter of the IFC and indicators of systolic (LVEF (r=-0.42, p=0.002), the rate of systolic shortening of myocardial fibers (r=-0.54, p=0.00005), % systolic shortening of the IVS (r=-0.29, p=0.04), % systolic shortening of the fibers of the left ventricle (r=-0.37, p=0.007)) and LV diastolic function (time of acceleration of early transmitral diastolic flow (r=- 0.52, p=0.0001), peak velocity of transmitral diastolic flow (r=-0.49, p=0.0003) and its gradient (r=-0.48, p=0.0004)).
Our information about the pathological expansion of the IFC in DST is consistent with the data of other authors [1; 5]. At the same time, we have not found any information in the literature about the narrowing of ROS during DST. The decrease in ROS in our patients can be explained by a decrease in LV volume in diastole (r=0.46, p=0.0003), a decrease in LV SV (r=0.51, p=0.00005). In work [6], individuals with MVP had a significantly larger diameter of the aortic annulus fibrosus compared to controls (30.4 ± 0.1 versus 29.5 ± 0.1 mm). This may be due to the fact that the patients in this study were older (mean age 37.9 ± 0.3 years) and did not differ from the control group in terms of BMI (23.7 ± 0.1 kg/m2). In our sample, the average age of patients with DST was 25.9±6.8 years, the average age of the control group was 25.2±5.4 years; patients differed significantly in BMI. In the group with CTD, the median BMI was 19.3 kg/m2 (quartiles 17.7-21.2), in the control group, respectively, 22.8 (21.2-25.6) kg/m2 (p<0.00001).
In our sample, significant correlations of ROS diameter with BMI (r=0.38, p=0.003), as well as with the Pinier index (r=-0.38, p=0.005) were obtained. Thus, it is more likely that the smaller diameter of the ROS is associated with the smaller body size of patients with CTD and the smaller size of the heart chambers. However, with smaller heart chamber sizes in patients with CTD, the diameters of the TFC and PFC did not differ between the groups.
Literature:
- Analysis of left ventricular myocardial deformation during mitral valve prolapse / E.G. Maleev [et al.] // Vestn. St. Petersburg. honey. acad. postgraduate education. – 2011. – No. 2. – P. 134-142.
- Achievements and prospects for studying the phase structure of the cardiac cycle using Doppler echocardiography. Possible applications in pediatrics [Electronic resource]. – Access mode: https://crawfordcountyredcross.org/kardiologiya/dostizgeniya_i_ perspektivy. html. – [Date of access: 02.21.12].
- Nechaeva G.I. Connective tissue dysplasia: terminology, diagnosis, patient management tactics / G.I. Nechaeva, I.A. Viktorova. – Omsk: LLC Printing House BLANKOM, 2007. – 188 p.
- Rybakova M.K. Practical guide to ultrasound diagnostics. Echocardiography / M.K. Rybakova, M.N. Alekhin, V.V. Mitkov. – M.: Vidar-M, 2008. – 512 p.
- Yakovlev V.M. Connective tissue dysplasia of the mitral valve / V.M. Yakovlev, R.S. Karpov, E.V. Shvetsova. – Tomsk: Sib. Publishing house House, 2004. – 144 p.
- Isolated mitral valve prolapse is an independent predictor of aortic root size in a general population / JR Matos-Souza // Eur. J. Echocardiogr. – 2010. – Vol. 11, No. 3. – P. 302-305.
Fibrosis of the heart and liver. Renin-angiotensin-aldosterone system and fibrosis
00:00
Drapkina Oksana Mikhailovna , executive director of the Internet Session, secretary of the interdepartmental council on therapy of the Russian Academy of Medical Sciences:
— I will give the next lecture. It will be called “Fibrosis of the heart and liver: what is common.”
Dear colleagues, the topic of fibrosis, in my opinion, is very relevant. It gives us the opportunity, if possible, to make our tissues (connective, parenchymal, muscle cells, any other tissue) live longer. Fibrosis is the natural aging of the body. Fibrosis of any organ (I want to emphasize again), any cell (it could be an endothelial cell, a vascular cell, a liver cell). The mechanisms of fibrosis development are largely similar.
Today I will try to begin our educational course, which we hope to make continuous. This problem will involve more than just cardiologists. Hepatologists will definitely be involved. In particular, Professor Pavlov, who works in our clinic. He deals with the liver problem very seriously. Pulmonologists and so on.
My task today is to talk about possible parallels between the formation of fibrosis of the heart and liver. Are there such parallels or are these fictitious attempts to connect different processes together?
I would like to pay attention to the role of the renin-angiotensin-aldosterone system in the pathogenesis of fibrosis formation. We have already seen today this ubiquitous angiotensin-2, which interacts with angiotensin type 1 receptors. It will identify all the adverse effects (in particular, by affecting insulin resistance, it increases the pressure of obese hypertensive patients).
Now let's look at it from a slightly different angle. From the perspective of fibrosis development.
What it is. Definition. Fibrosis is a thickening of connective tissue with the appearance of scar changes. We are very familiar with areas of fibrosis after, for example, a myocardial infarction.
These scar changes can occur in various organs. They arise, as a rule, at the site of chronic inflammation: atrophy or dystrophy.
A molecular definition can be given. We can say that fibrosis is the excessive accumulation of fibroblasts and extracellular matrix proteins, including collagen, in tissues.
Today, within 20 minutes, we must trace the fate of collagen, which is the main structural unit of tissue fibrosis.
The history of the study of the issue dates back to 1872, when William Gull and Henry Sutton described arteriocapillary fibrosis, which develops in kidney disease.
Nobel laureates have already worked in the 1930s of the twentieth century. They worked to uncover the structure of collagen. It was revealed.
03:22
In the 1960s, John Ross and Eugene Braunwald began to study the effect of angiotensin on cardiac function. Then the first works appeared on the relationship between diabetes mellitus and myocardial fibrosis. Or a hypertrophied left ventricle due to arterial hypertension and myocardial fibrosis.
Previously, Georgy Fedorovich Lang , the indisputable authority of the Russian medical school, had already found “gentle fibrosis” (as he said) in the hearts of patients suffering from arterial hypertension.
More recently, quantitative and qualitative assessment of fibrosis has become possible. Of course, we strive to develop more and more non-invasive methods in our diagnostic capabilities. Of course, liver biopsy and myocardial biopsy are not always available.
Liver biopsy is available, but myocardial biopsy is difficult. Now they are struggling with which method will be comparable in its sensitivity and specificity to the morphological one.
The main characters are whom we should know by sight when talking about fibrosis. These are fibroblasts, collagen. And here we are interested in collagen of the first and third types. These are components of the extracellular matrix. We will mainly talk about proteoglycans here.
The matrix metalloproteinases themselves and their inhibitors (that is, the ratio of matrix metalloproteinases that are responsible for collagen degradation and matrix metalloproteinase inhibitors) will be targets that can be targeted with various therapeutic agents in order to either increase collagen synthesis or have a greater effect on its degradation.
Profibrotic markers. The most famous are, of course, TGF beta (growth marker).
05:28
What is collagen? Collagen is a triple helix. This triple helix of collagen makes up 1/3 of all the proteins in the human body. It is formed by protein chains. When these triple helices of collagen, which are extended by protein chains, converge and stitch together, rod-shaped collagen molecules are formed. They form very strong, inextensible collagen fibrils. Their strength is comparable to that of steel.
You understand how difficult it is to reverse fibrosis, for example. In my opinion, it is better to do everything possible to prevent fibrosis from forming.
How does collagen synthesis occur? Collagen synthesis is a process that requires cellular and extracellular synthesis. The collagen precursor is synthesized on ribosomes on the surface of the endoplasmic reticulum. It's called preprocollagen. Post-tranquilizer modification then occurs in the Golgi complex. Some signal peptides are cleaved off.
Procollagen appears, which still carries a long propeptide at one end. This is followed by hydroxylation and glycosylation. Oxidation of cystoine residues leads to the formation of intermolecular disulfide bonds.
The very last stage is condensation with the formation of intra- and intermolecular bonds. When all these stages are completed, we get collagen.
(Slide show).
Section of the left ventricular myocardium of any patient. It is important that the connective tissue framework of the myocardium consists of three main components: epimysium, endomysium, perimysium. The endomysium surrounds a large muscle fiber. These are dense bundles of collagen that make up the connective tissue layer surrounding the entire myocardium. Epimysiums are part of the epicardium and endocardium.
Endomysiums are collagen fibers that surround each muscle fiber and form a mesh sheath. Around these muscle fibers there are also capillaries. This is also very important. Interpersonal relationship between muscle fiber and capillary.
08:48
Endomysium and epimysium connect perimysium. The role of perimysium is extremely important in relaxation processes. The perimysiums are rather thick bundles. They thicken, shorten during systole and lengthen, almost turning into a continuous ribbon during diastole. The more elastic the perimysium is, the more elastic our heart will be.
Thus, in the process of myocardial fibrosis, which we will talk more about today, these three bundles play a great importance. Epimysium, endomysium, perimysium.
(Slide show).
Ultrastructural level. On the left is an electron micrograph of a fragment of the left atrium. We see how collagen fibers are located between the fibroblast and the capillary. Most likely, we are talking about endomysium here.
The adjacent image shows how the cardiomyocyte is separated from the capillary by a thin plate of fibroblast growth. If the fibroblast is activated (we will recall once again below what agents it can be activated), this will lead to the fact that the fibroblast process will gradually be replaced by connective tissue.
Let's make the complex simple. Let's ask ourselves: what is the process of collagen formation? Or what fibrosis is. Fibrosis is the predominance of collagen synthesis over its breakdown.
Collagen synthesis and conversion requires lysyl oxidase activity. Then metalloproteinase comes into play. Metalloproteinase inhibitors will reduce the effect of the latter metalloproteinases. Collagen degradation occurs.
We must not confuse what we see (fibroblast). We must understand that this is not just a predominance of collagen synthesis over its breakdown. This is also the process of collagen structuring. Only structured collagen is important in the development of fibrosis, and not just bundles of collagen that will be scattered throughout the myocardium.
11:27
We know very well the different types of fibrosis, which indicate which path the tissue will take: apoptosis or necrosis. Normal tissue consists of cardiomyocytes that are connected to each other end to end.
If myocardial infarction suddenly occurs and reparative fibrosis forms at the site of the scar, then normal cells begin to alternate with cells - sections of connective tissue. Such reparative fibrosis occurs at the site of dystrophic changes. Here, arttrophy and tissue degeneration underlie this type of fibrosis.
Reactive fibrosis is something we see much more often. These are all hypertensive patients (many, in any case) with a long history of arterial hypertension (AH), with poorly treated hypertension. Reactive fibrosis. Here there is an increase in the intercellular matrix. Collagen bundles become thicker.
Vladimir Leonidovich spoke today about arrhythmias. My deep conviction is that, apparently, fibrosis processes are to blame for arrhythmias (especially supraventricular ones). In particular, perhaps, in the atria.
We will have 20 minutes of discussion. I think everyone can have their say on this matter.
Many experimental studies have proven that fibrosis can serve as the basis for the development of tachyarrhythmia. The most arrhythmogenic zone is the zone of the mouth of the pulmonary veins. They try to limit this zone during minor surgical interventions for rhythm disturbances.
(Slide show).
With tachyarrhythmias, the slices take on this shape. White is areas of fibrosis. The more fibrosis, the more prepared such an atrium will be for takiarrhythmia.
13:51
Here we see disorganized bundles of myocardial sleeves. They very densely envelop the mouths of the pulmonary veins. Accordingly, there are even works that suggest that the manifestations of fibrosis and its prevalence and localization may have prognostic significance for the choice of tactics for managing a patient with arrhythmia.
What affects fibrosis? What is beneficial for us, on the contrary, is to stimulate in order to prevent the development of fibrosis. We are interested in fibrogenesis stimulators and inhibitors.
Angiotensin-2 is a fibrogenesis stimulator. There are intimate connections between angiotensin-2 and TGF beta. These connections are not fully disclosed. Today we will try to talk about them again.
Other growth factors. The second character we know well is aldosterone. The more angiotensin-2 and aldosterone in our patient's plasma or tissue system, the greater the potential for him to have fibrosis of the heart and (as I will show later) liver.
Endothelins, catecholamines, adhesion molecules, galectin-3. There is now a lot of work being done on the role and specificity of galectin-3, which increases during fibrosis. On the other hand, an increase in the synthesis of nitric oxide, an increase in the activity of natriuretic peptides, bradykinin, and prostaglandins leads to the fact that fibrosis in such patients (if the activity of these agents is high) will occur to a lesser extent.
Well-known hormones (growth hormone, thyroxine, angiotensin-2 and aldosterone). If we compare their effect on several stages in the development of fibrosis (collagen synthesis, collagen degradation and collagen accumulation), note that growth hormones and thyroxine affect collagen synthesis and degradation. In these practically equally directed processes, conditions are not created for collagen accumulation to occur and connective tissue to accumulate.
On the contrary, the influence of angiotensin-2 and aldosterone predominates on collagen synthesis. Angiotensin also reduces collagen degradation, which has a very pronounced effect on collagen accumulation. Aldosterone primarily acts on collagen synthesis without affecting collagen degradation. Accordingly, accumulation also occurs, but to a much lesser extent.
16:59
(Slide show).
A well-known scheme was published in 2008. Cardiomyocytes and fibroblasts. Muscle fiber contains cardiomyocytes and fibroblasts. Cardiomyocytes are shown in pink. Fibroblasts are depicted as ugly shapes in blue with a nucleus.
In response to mechanical distension (for example, increased pressure in the left ventricle because systemic arterial pressure (BP) rises or for some other reason), the pressure inside the ventricle increases. According to the pressure gradient, there will be an increase in the system, for example, the left atrium. This results in mechanical stretch, which stimulates TGF beta activity.
At the same time, angiotensin-2, influencing the same mechanisms and mechanisms that occur in fibroblasts, also activates TGF beta activity. All this leads to an increase in the organization of collagen and other proteins of the intercellular matrix and fibrosis occurs.
What can affect fibrosis. Something that can block the renin-angiotensin-aldosterone system. There have been many novel models that have shown that angiotensin-converting enzyme inhibitors lead to a significant reduction in fibrosis.
Today I will dwell more on Lisinopril, since I also added liver fibrosis to this fibrosis.
If we talk about liver fibrosis, the mechanism was similar. Again we see TGF beta in the arena, which will be activated by angiotensin-2. You have the right to ask me: where does angiotensin-2 come from in the liver? Liver tissue (like any tissue) is equipped with blood vessels. There are many vessels. It is with the processes of changes in blood vessels that the processes of fibrogenesis begin to occur in the liver. I will show this a little later.
There are certain parallels in the development of fibrosis of the liver and heart. These parallels lie in the same clinical predictors of the development of fibrosis and cirrhosis of the liver and cirrhosis and fibrosis of the heart.
Firstly, this is age. Secondly, it is an increased body mass index. Thirdly, these are comorbid conditions such as, for example, insulin resistance and diabetes mellitus. Today we also talked about how angiotensin-2 acts on insulin resistance. This is a change in transaminase levels and an increase in triglyceride levels.
20:08
In the heart, reactive fibrosis is observed in hypertension and abdominal obesity. Epicardial fat, which is visceral fat and a springboard for the secretion of adipokines and faster myocardial fibrosis, which is also initially associated with impaired diastole. This means fibroformation, which increases in such patients.
Thus, we can say with some confidence that epicardial fat is a new marker of cardiovascular disease (CVD). It correlates with both visceral fat and subclinical atherosclerosis, the outcome of acute coronary syndrome (ACS) and, finally, the diagnosis of metabolic syndrome.
A few more words about fibrosis of the liver and heart. Common pathophysiology. We need to focus on the very interesting agent galectin-3. Now there is a lot of work that shows that galectin-3 increases in liver fibrosis.
Personal attention is given to an oral caspase inhibitor and an apoptosis inhibitor. These studies are still in progress.
I promised to prove that the liver and blood vessels are by no means mutually exclusive. The liver is permeated with blood vessels. It is with the calillarization of the sinusoid that the processes of fibroformation begin. If normally the basement membrane is a membrane with pronounced spaces of Disse (that is, fenestrations are detected between the sinusoids), then with mechanical damage the sinusoids lose fenestrations. Exchange with the capillary and blood flow changes. This is how fibrosis forms.
Otherwise, ACE inhibitors have something to act on in the liver of such a patient.
The last component is vascular fibrosis. Vascular fibrosis is a process that is obligatory in an elderly person. It increases with age. This is the main reason for the increase and stiffness in hypertension. It is possible to draw a logical connection that a stable increase in blood pressure leads to the activation of fibroblasts and a decrease in the activity of metalloproteinases.
12:47
The only thing that is not clear is what initiates the process of fibrosis. It is possible that the role of immune cells or immune cells that infiltrate the vascular wall perform this unpleasant role.
Perivascular fibrosis due to diabetes mellitus. There are a lot of such patients.
Dear colleagues, remember your workday yesterday, your workday today. You probably remember patients with diabetes who come with chest pain. On coronary angiography, their coronary arteries are clear. What's the matter?
The fact is that this anginal status may be a consequence of extravasal compression of the coronary arteries, because glucose promotes the formation of collagen “crosslinks”. In patients with diabetes mellitus, perivascular fibrosis is very pronounced.
The effects of blockers that inhibit the renin-angiotensin-aldosterone system are pathogenetically justified for the prevention of fibrosis, both in the heart and in the liver. They act on TGF beta and on the activity of some metalloproteinases. For example, metalloproteinases of the first type.
All this confirms the term: what is good for the heart is also good for the liver. There is no longer any doubt about this. I'll touch on this briefly.
Non-alcoholic fatty liver disease. It would seem a completely safe, harmless state. But not so harmless. These patients have the opportunity to die from CVD many times more often than without such suffering.
Increased risk of carotid atherosclerosis. The kinetics of the thrombus changes. Non-alcoholic fatty liver disease is a predictor of CVD, independent of other risk factors. The ways of applying and correcting this condition, which at first glance seems harmless, are changing.
If we are talking about fibrosis, then ACE inhibitors, statins, and renin-angiotensin receptor blockers must be used in such a patient.
25:14
What to choose. What to prescribe to an obese hypertensive patient from the perspective that fibrosis exists in both the heart and liver. Probably, drugs that do not require additional metabolization in the liver and that circulate in the plasma link of the renin-angiotensin-aldosterone system come to the fore here.
From this point of view, Lisinopril has proven itself to be very good. We use the drug “Diroton” in our practice. When you ask doctors if there is an original drug, Lisinopril, they all answer that it is Diroton. He has proven himself so well. Its metabolic neutrality and lack of transformation in the liver are captivating.
Very often, a patient who has fibrosis (this is a high-risk patient) needs combination therapy. Today we talked a lot about this combination (extremely interesting, metabolically neutral). Calcium antagonists and things that block the renin-angiotensin-aldosterone system.
There is a combination “Equator”, which consists of “Lisinopril” and the beloved “Amlodipine”, which we also use in patients.
In this regard, I would like to recall and refer to the results of a study that we conducted earlier. We tried to see what Lisinopril would do in a patient with concomitant liver pathology.
We took patients with hypertension (mild and moderate). There were 25 patients. We looked after them for 12 weeks. The dose of Lisinopril was titrated from 5 mg to 20 mg per day. It turned out that during a routine examination...
There were 4 visits, at each visit the patient’s clinical status was examined in detail: electrocardiogram, echocardiogram, dose titration and blood pressure monitoring were performed. The patients were distributed as follows. These were mainly patients with moderate hypertension. The experience wasn't very long. Experience prevailed from one to five years.
According to concomitant liver disease, patients were distributed as follows: mainly included patients who had alcoholic liver damage. At the third visit, a significant decrease in blood pressure levels was already noted with Lisinopril. This was all accompanied by a decrease in the levels of transaminases, glutamine transpeptidase and alkaline phosphatase.
All patients demonstrated excellent tolerability. One patient discontinued lisinopril in this study due to headache.
28:14
So, what can we say about fibrosis of the liver and heart. First, this problem is urgent. Secondly, these two processes in two completely different organs (vessels should be added here) have many similar indicators. This makes it possible and necessary to use drugs that have proven themselves in terms of their effects on the cardiovascular system and liver.
Principle: What is good for the heart is also good for the liver.
The last conclusion: the reduction of fibrosis is a process that is now very much developing. We need to read, watch, understand that such drugs as Relaxin, statins, inhibitors of the renin-angiotensin-aldosterone system. Good experience has been accumulated with Diroton. Destroyer of cross-links of proteins. Oxidase inhibitors, which interfere with collagen organization. Phospholipase D inhibitors. Genetic therapy. Especially micro-RNKs. I think that this will help solve the problem of fibrosis in the next 100 years.
Thank you for your attention.