It is customary to talk about lipid metabolism disorders in the body if there is an imbalance between the levels of high- and low-density lipoprotein cholesterol, as well as triglycerides. This causes many health problems and leads to the development of serious diseases, including cardiovascular disasters - myocardial infarction and stroke.
The head of the Department of Cardiology and Rheumatology of BelMAPO, Doctor of Medical Sciences, Professor Andrey Pristrom told the journalist of the information portal “Healthy People” about the causes of lipid metabolism disorders, what it entails and how to diagnose the pathology .
Symptoms, causes, diagnosis and treatment of lipid metabolism disorders
The increase in the content of fat-like substances - lipids - in the blood is influenced by three main factors: excess intake of fat into the body from food, impaired excretion and increased synthesis in the body.
Depending on the mechanism of development of the disease, the causes of dyslipidemia are different.
Pathology can be:
- primary (due to hereditary factors);
- secondary (as a result of complications of any diseases);
- nutritional (if the human diet is oversaturated with animal fats).
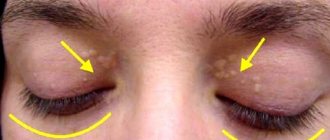
An anomaly is often recognized after the fact, when problems with the cardiovascular system arise, since lipid metabolism disorders are most often asymptomatic. But in some patients, due to high levels of LDL, clouding of the cornea occurs, and xanthomas appear on the elbows and knees, and on the soles. With very high triglyceride levels (exceeding 11.3 mmol/l), pancreatitis develops and xanthomatous rashes are observed on the body.
Mucopolysaccharidoses
A group of hereditary connective tissue diseases characterized by combined damage to the musculoskeletal system, internal organs, eyes and nervous system. Increased excretion of mucopolysaccharides in the urine and their accumulation in various organs and tissues. There are nine types of the disease. Here are some of them.
47. MSSI syndrome. The enzyme iduronidase is affected. Mental retardation is pronounced. Prenatal diagnosis is possible. Various somatic anomalies are detected. The type of hereditary transmission is autosomal recessive.
Diseases 48–52 are characterized by similar somatic disorders: varying degrees of bone damage, hepatosplenomegaly, limited joint mobility, etc.
48. Gunther's disease II. The enzyme iduronate sulfase is affected. Mental retardation is pronounced. Somatic abnormalities are observed. Prenatal diagnosis is possible. The type of hereditary transmission is recessive, linked to chromosome X.
49. Sanfilippo III syndrome. Mental retardation is combined with such anomalies as thickening of the skull bones, enlarged liver, and deformation of the lumbar vertebrae. Prenatal diagnosis is possible. The type of hereditary transmission is autosomal recessive. Various enzymes are affected (phosphatases types A-D).
50. Morquio's disease IV. The enzyme N-acetylgalactosamine is affected. There are no data on mental retardation. Prenatal diagnosis is possible. The type of inheritance is autosomal recessive.
51. Maroteaux-Lami disease IV. The enzyme arylsulfatase B is affected. Mental retardation does not always occur. Prenatal diagnosis is possible. The type of inheritance is autosomal recessive.
52. Gargoilism (the name comes from the images of chimeras that decorated ancient drainpipes). Other names: Gurler disease, Johnny McL disease. (name of the first patient), multiple dazostosis, Pfaundler's disease. The main symptoms are: cognitive deficit, chondroosteodystrophy, hepatosplenomegaly, corneal opacification. A large head, an ugly structure of the facial part of the skull, dwarf stature, and increasing blindness are typical. The type of hereditary transmission is autosomal recessive. Prenatal diagnosis of the disease is possible.
Classification of lipid metabolism disorders
Today, the Fredrickson classification of dyslipidemias is considered the main one:
- The cause of type 1 dyslipidemia is enzyme deficiency. Violations of this type are quite rare.
- Dyslipidemia 2a occurs due to mutations in genes and is one of the most common types of lipid metabolism disorders.
- Dyslipidemia 2b also occurs quite often and can be either hereditary or combined (the cause may be previous diseases and a diet oversaturated with animal fats).
- Type 3 lipid metabolism disorders are characterized by an increase in triglycerides and LDL in the blood.
- Type 4 hyperlipidemia, characterized by an increase in VLDL, is of endogenous origin.
- Dyslipidemia type 5 occurs when the level of cholinomicrons in the blood increases and also refers to hereditary disorders.
Hereditary diseases of lipid metabolism
Hereditary diseases of lipid metabolism are a large group of diseases that arise as a result of disruption of one of the stages of synthesis, transport and degradation of lipoproteins, which include all the main plasma lipids - triglycerides (TG), phospholipids, cholesterol (C) and free fatty acids. Blood plasma lipids are most often found in combination with various apoproteins, forming lipoproteins. To date, the 8 main apoproteins have been best studied - Apo-A 1,2,4, Apo-B, Apo-C 1,2,3 and Apo-E. Some apoproteins are integral parts of certain lipoproteins, while others can migrate from one lipoprotein to another.
There are four main classes of plasma lipoproteins:
1) chylomicrons (CM) - are formed in the cells of the intestinal mucosa and are carriers of dietary TGs;
2) very low density lipoproteins (VLDL) - are formed in the liver and are used to export TG to other tissues;
3) low-density lipoproteins (LDL) - are the end products of VLDL catabolism and carriers of cholesterol into cells;
4) high-density lipoproteins (HDL) - metabolize cholesterol and VLDL and transport cholesterol from cell membranes to the liver.
A typical lipoprotein consists of a lipid core formed by triglycerides and cholesterol esters and an outer layer composed of phospholipids, cholesterol and apoproteins.
Normal lipid metabolism occurs as follows. VLDL synthesized in the intestines and liver enter the bloodstream and transport TG (triglycerides) to fat cells and other tissues. After the separation of TG in fat cells, cholesterol re-enters the liver, and VLDL is converted into LDL, enriched with cholesterol and containing Apo-B and Apo-E. LDL remains in the bloodstream for 2.5 days and serves as cholesterol transporters to various cells. The entry of cholesterol into cells is carried out using LDL receptors, the number and functions of which are under genetic control. Cell receptors are grouped in special areas of the cell membrane, immersed like a crater in the cytoplasm. After the receptor interacts with LDL, the portion of the membrane containing the receptors is drawn into the cell and forms a bordered vesicle. In the cytoplasm, LDL is separated from the receptor and enters lysosomes, and the receptors are returned to the cell membrane. The release of cholesterol from LDL is carried out under the action of several enzymes. The return delivery of cholesterol to the cells of the liver and small intestine is carried out by HDL also using the mechanism of receptor endocytosis.
To date, more than 420 different mutations have been described, which, depending on the impact, can be grouped into 5 classes:
1) leading to the cessation of receptor synthesis;
2) disrupting the migration of the receptor from the endoplasmic reticulum to the Golgi apparatus;
3) changing the process of binding the receptor to LDL;
4) disrupting receptor clustering on cell membranes;
5) disrupting the process of LDL cleavage in the cell and transport of the receptor to the cell membrane.
The main stages of cholesterol metabolism in the body are under complex genetic control. Disruption of various stages of this metabolic pathway as a result of mutations can lead to a delay in the removal of cholesterol from the body and the emergence of various types of hyperlipidemia, the development of atherosclerosis of the blood vessels of the heart and brain. In recent years, it has been possible to decipher the molecular genetic mechanisms of some monogenic diseases caused by lipid metabolism disorders.
Lipoidoses are a large group of hereditary abnormalities of lipid metabolism, the common feature of which is a high level of lipids in the blood plasma (plasmatic lipoidoses) or the accumulation of lipid metabolism metabolites inside cells (intracellular lipoidoses). Lipoidosis includes diseases of lipoprotein metabolism. The most common forms of lipoidosis are Gaucher and Niemann-Pick, Tay-Sachs diseases, which are classified as so-called “storage diseases”, the clinical differentiation of which is extremely difficult.
Gaucher disease belongs to sphingolipidoses - diseases of lipid accumulation; is caused by a defect in the gene responsible for the synthesis of the lysosomal hydrolytic enzyme beta glucocerebrosidase (beta glycosidase).
The disease is inherited in an autosomal recessive manner.
Defect and deficiency of this enzyme lead to impaired utilization of lipids - glucocerebrosides and their accumulation in macrophages, mainly of the bone marrow, spleen, and liver. The diagnosis is established by the detection of specific Gaucher cells (lymphocyte-like nucleus, eccentrically located, and very wide light cytoplasm with slightly noticeable circular striations) in the puncture of the spleen (puncture can only be done in a hospital) or in the bone marrow.
There are three types of Gaucher disease. Type 1 (benign) – adult type. It is 30 times more common among Jews (Western European Ashkenazi group). There are no neurological disorders, visceral changes are associated mainly with hematopoietic organs, enlarged spleen, hypersplenism, and destruction of bone tissue.
Type 2 – infantile. It is a malignant form of the process with severe neurological disorders that manifest themselves in newborns and lead to death in the first 2 years of life.
Type 3 – juvenile. Differs in the variability of visceral and neurological disorders; in its course it is less malignant than type 2. The variety of forms of Gaucher disease is due to the heterogeneity of mutations of the beta glycosidase gene.
Niemann-Pick disease (sphingomyelinosis). It belongs to the group of thesaurismoses (storage diseases) and is characterized by severe damage to the nervous system and a delay in the overall development of the child.
Inheritance of the disease occurs in an autosomal recessive manner. Boys and girls get sick equally often. Consanguinity between parents is often noted, and there are familial cases of Niemann-Pick disease.
The pathogenesis of the disease is associated with a violation of the synthesis of sphingomyelin, which contains an excess amount of some fatty acids and a deficiency of others. Perhaps, disturbances in the processes of sphingomyelin breakdown are of some importance, which leads to its excessive accumulation in the reticuloendothelial cells of various organs and tissues. Microscopically, Pick cells are found in the spleen, liver, kidneys, adrenal glands, lymph nodes, bone marrow and some other organs. These are quite large cells, ranging in size from 20 to 50 microns and containing one or many nuclei. The protoplasm of the cells contains vacuoles, which give the cells their characteristic foamy appearance. Cell foam is formed due to the dissolution of lipid substances during fixation of the drug. Histological changes are found in ganglion cells of the brain and retina. The amount of sphingomyelin in various organs increases sharply. In 1961, the following classification of the disease was proposed:
— Niemann-Pick disease, type A: classic infantile;
— Niemann-Pick disease, type B: visceral;
— Niemann-Pick disease, type C: non-acute teenage
— Niemann-Pick disease, type D: Nova Scotia;
However, today, when the genetic nature of the disease is understood, the disorder is classified as follows:
- Niemann-Pick disease associated with the SMPD1 gene, which includes types A and B;
- Niemann-Pick disease, type C, which includes types C1 and C2. (Type D results from a mutation in the same gene as type C1).
Clinically, Niemann-Pick disease usually manifests itself in infancy, often in the first six months. There are isolated descriptions of the occurrence of clinical signs of the disease in older children and even young adults. The initial symptoms of the disease are loss of appetite, sudden weight loss of the child, and delayed psychophysical development. The patient's abdomen increases significantly due to hepatosplenomegaly (enlarged spleen and liver). Bronchopneumonia often occurs. Lymph glands are enlarged in most cases. The skin looks waxy, shiny, and areas of pigmentation are found. Neurologically, in the initial stages of the disease, motor disorders of a central nature are detected: paresis of the limbs, increased muscle tone and tendon reflexes, pyramidal signs. In later stages, muscle hypotonia and absence of tendon reflexes are characteristic. Idiocy, blindness and deafness develop. In the fundus of many patients, atrophy of the optic nerve nipples and an oval-shaped cherry-red spot in the macular area may be detected. Niemann-Pick disease has a malignant course. Most children die in the first 2 years of life from pulmonary heart failure and intercurrent infections.
Tay-Sachs disease is an inherited neurometabolic disease associated with the accumulation of GM2 ganglioside in the brain. Gangliosides contain one or more sialic acid residues.
Autosomal recessive type of inheritance. Tay-Sachs disease is caused by mutational lesions of the hexosaminidase (HEXA) gene, which controls the synthesis of the alpha subunit of hexosaminidase A. Hexosaminidase A is a lysosomal enzyme that catabolizes the catabolism of GM2 ganglioside.
There are three forms of Tay-Sachs disease:
Child form - six months after birth, children experience a progressive deterioration in physical abilities and mental abilities: blindness, deafness, and loss of the ability to swallow are observed. As a result of muscle atrophy, paralysis develops. Death occurs before the age of 3-4 years.
Adolescent form - motor-cognitive problems, dysphagia (impaired swallowing), dysarthria, (speech disorders), ataxia (unsteadiness of gait), spasticity (contractures and paralysis) develop. Death occurs before the age of 15-16 years.
Adult form - occurs between the ages of 25 and 30 years. It is characterized by symptoms of progressive deterioration of neurological functions: impaired and unsteady gait, swallowing and speech disorders, decreased cognitive skills, spasticity, and the development of schizophrenia in the form of psychosis.
Fabry disease is a rare inherited X-linked recessive lysosomal storage disorder. Deficiency of the enzyme alpha-galactosidase A, which occurs due to a mutation, causes the accumulation of a glycolipid known as globotriaosylceramide (abbreviated GB3, GL-3) in blood vessels and other tissues and organs. This accumulation leads to disruption of their normal functioning.
Whole-body pain or localized pain in the extremities (known as acroparesthesia) or gastrointestinal (GI) tract is common in patients with Fabry disease. It is believed that acroparesthesia in the disease is associated with damage to the peripheral nerve fibers that transmit pain. Pain in the gastrointestinal tract is likely due to the accumulation of lipids in its small vessels, which in turn interferes with blood circulation (intestinal ischemia), causing pain. Kidney-related complications are a common and serious consequence of the disease. Kidney failure (uremia) may worsen over time. Proteinuria (which can cause foamy urine) is often the first sign of kidney damage. End-stage renal failure in men usually occurs between 30 and 40 years of age and is a common cause of death. Angiokeratomas (small, painless papules that can appear on any part of the body, but usually appear on the thighs, around the navel, buttocks, lower abdomen and groin) are also a common symptom. Anhidrosis (lack of sweating) is a common symptom, unlike hyperhidrosis (excessive sweating), which is much less common. The disease also causes eye damage, namely clouding of the cornea (known as keratopathy).
Hereditary deficiency of hepatic lipase, or hyper-a-triglyceridemia, is characterized by the accumulation of TG in the a-LP fraction. As a result of inhibition of reactions catalyzed by hepatic lipase in the blood. Manifestations: lipoid arc of the cornea, xanthomas, palmar striae, ischemic heart disease.
Hyperlipidemia is an increase in lipid levels in the blood or tissues caused by congenital or acquired metabolic disorders.
Genetic factors interact with environmental factors, including diet and medications. Inherited components are often polygenic and difficult to define, but three purely inherited disorders have been described—familial hypercholesterolemia (FH), familial hyperlipoproteinemia type III, and familial combined hyperlipidemia.
There are five types of hyperlipoproteinemia with specific symptoms characteristic of each type:
Type I is characterized by attacks of stomach pain, usually after eating fatty foods, as well as a general deterioration in health, loss of appetite and fever.
Type II is characterized by the appearance of dense formations on the Achilles tendons and tendons of the hands and feet
Type III may cause soft, inflamed sores to appear over the elbows and knees. Type IV is caused by overeating, obesity and diabetes.
Type V is characterized by abdominal pain (the most common symptom), yellow nodules on the skin and reddish-whitish blood vessels on the retina of the eyes
Hereditary hypercholesterolemia is an autosomal dominant, monogenic disease, the development of which is caused by a violation of the structure or function of receptors for low-density lipoproteins (LDL) on somatic cells (liver and others) and/or their number [5,13] (classical form of NHHS [5 ]), or disturbances in the structure of the molecule of apolipoproteins B-100 and C (apo B-100 and C, the protein components of lipoproteins).
Depending on the nature of the genetic defect, the disease is classified as receptor-negative, receptor-defective, or receptor-internalizing forms.
The main disorders are impaired catabolism of LDL by the liver and other tissues through LDL receptors that bind to Apo B. With pathology of LDL receptors, this causes an increase in the level of LDL in the blood and an increase in the circulation time of this type of lipoprotein, which ultimately leads to the formation of altered, modified forms of LDL, which are highly atherogenic. Homozygous forms of NHCS differ from heterozygous ones in the complete absence of LDL receptors on the surface of cells and removal of LDL from the bloodstream in this way, and longer circulation in the peripheral bloodstream as a result of these changes (capture of LDL by cells occurs in a non-receptor manner), whereas in heterozygous forms the number of receptors reduced by about half.
On the skin of patients with NHHS, pathognomatic signs of lipid metabolism disorders are observed: xanthomas and xanthelasmas (deposition of cholesterol esters in the thickness of the skin).
Diagnostic measures
Diagnostic procedures include a consultation with a physician, physical examination, medical history, and blood chemistry tests.
During a physical examination, the doctor notes the possible presence of xanthelasma, xanthomas, and lipoid arch of the cornea. Patients often experience high blood pressure. Auscultation (listening) and percussion (tapping) are not informative, since they are not accompanied by changes in dyslipidemia.
To identify inflammation and concomitant diseases, a laboratory test of blood and urine is prescribed. Genetic analysis identifies genes that carry hereditary information and are responsible for the development of type 2 dyslipidemia.
A biochemical blood test allows you to determine the level of total blood protein and sugar, uric acid and creatinine to detect concomitant organ damage. Immunological analysis determines the content of antibodies to cytomegalovirus and chlamydia, as well as the level of C-reactive protein.
But the main method for diagnosing dyslipidemia is a lipidogram - a blood test for fat-like substances, lipids. When measuring the lipid spectrum
The levels of triglycerides, total cholesterol, LDL and HDL are determined.
The studies are carried out on an empty stomach, in NEARMEDIC's own laboratory.
Treatment of dyslipidemia is prescribed after a complete diagnosis, accurate diagnosis and determination of the cause of the pathology.
Oligosaccharide and glucoprotein dysmetabolism
53. Cellular inclusion disease. Glycoprotein enzymes, N-acetylglucosaminylphosphotransferase, are affected. Severe mental retardation, and in somatic terms - hepatomegaly, disorders of the skeletal system, swelling of the gums. Prenatal diagnosis is possible. The type of inheritance is autosomal recessive.
54. Mannosidosis. The enzyme mannosidase is affected. Severe mental retardation. Somatically: hepatomegaly, disorders of the skeletal system, roughening of the face. Prenatal diagnosis is possible. The type of inheritance is autosomal recessive.
55. Fucosidosis. The enzyme fucosidase is affected. Marked mental retardation. Somatically (see paragraph 56). Prenatal diagnosis is possible. The type of inheritance is autosomal recessive.
Treatment of dyslipidemia
Treatment of dyslipidemia is complex and includes:
- Drug therapy - fibrates, vitamins, statins and other drugs that correct lipid metabolism disorders;
- Non-drug treatment - weight normalization through fractional meals, dosed physical activity, limiting alcohol and smoking, and stressful situations.
- Diet therapy - foods rich in dietary fiber and vitamins are recommended (vegetables, cereals, fruits, beans, low-fat lactic acid products); fatty and fried meats are not allowed.
If lipid imbalance is a secondary pathology resulting from exposure to negative factors or any disease, NEARMEDIC cardiologists prescribe therapy aimed at timely detection and treatment of the underlying disease.
Dyslipidemia develops over years and requires equally long-term treatment. You can prevent further disturbances in lipid metabolism by strictly following the recommendations of doctors: move more, watch your weight, quit bad habits.
Contact your doctors on time!
In the early stages, stopping the pathological process is much easier. Timely therapy, elimination of risk factors and disciplined implementation of doctors’ recommendations significantly prolong and improve the lives of patients
Contact our clinics, do not delay your visit to the doctor. You will be consulted by experienced doctors, and you will undergo an expert examination using high-tech diagnostic equipment. Based on the results obtained, the cardiologist will prescribe competent treatment for dyslipidemia and recommend preventive measures.
To make an appointment with a cardiologist, call or fill out a request on the website.
Our clinics in St. Petersburg
Structural subdivision of Polikarpov Alley Polikarpov 6k2 Primorsky district
- Pionerskaya
- Specific
- Commandant's
Structural subdivision of Zhukov Marshal Zhukov Ave. 28k2 Kirovsky district
- Avtovo
- Avenue of Veterans
- Leninsky Prospekt
Structural subdivision Devyatkino Okhtinskaya alley 18 Vsevolozhsk district
- Devyatkino
- Civil Prospect
- Academic
For detailed information and to make an appointment, you can call +7 (812) 640-55-25
Make an appointment
There are 5 types of lipoproteins:
- Chylomicrons are synthesized by intestinal cells, after which they enter the lymphatic vessels and then into the blood. They are a transport form of triglycerides, cholesterol and exogenous fatty acids.
- Very low density lipoproteins . They are formed in the liver and contain up to 15 percent of total cholesterol. Very low density lipoproteins can be transformed into small particles, most of which are excreted in the liver.
- Intermediate density lipoproteins are an intermediate phase of very low density lipoproteins.
- Low-density lipoproteins (LDL) contain 60-70% cholesterol and are considered the main carriers of cholesterol to tissues. The role of low-density lipoproteins has been most studied in the process of atherogenesis. It produces smaller and denser particles. This type of disorder often occurs in diabetes mellitus.
- High-density lipoproteins (HDL) contain about 20-30% cholesterol. They play a very important role in the reverse transport of cholesterol from tissues to the liver. Experts have established a connection between a decrease in the concentration of HDL in the blood and the risk of coronary heart disease.