О нарушениях липидного обмена в организме принято говорить, если наблюдается дисбаланс между уровнем холестерина липопротеидов высокой, низкой плотности, а также триглицеридов. Это становится причиной многих проблем со здоровьем и влечет за собой развитие серьезных заболеваний, в том числе сердечно-сосудистых катастроф – инфаркта миокарда и инсульта.
О причинах нарушения липидного обмена, чем это чревато и как диагностировать патологию журналисту информационного портала «Здоровые люди» рассказал заведующий кафедрой кардиологии и ревматологии БелМАПО, д.м.н., профессор Андрей Пристром.
Симптомы, причины, диагностика и лечение нарушения липидного обмена
На повышение содержания в крови жироподобных веществ – липидов, оказывают влияние три основных фактора: избыточное поступление жира в организм с пищей, нарушение выведения и повышение синтеза в организме.
В зависимости от механизма развития заболевания, причины дислипидемии различны.
Патология может быть:
- первичной (обусловленной наследственными факторами);
- вторичной (как результат осложнения каких-либо заболеваний);
- алиментарной (если рацион человека перенасыщен животными жирами).
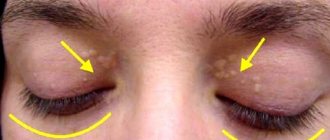
Распознают аномалию часто по факту, при возникновении проблем с сердечно-сосудистой системой, так как нарушение обмена липидов чаще всего протекает бессимптомно. Но у некоторых пациентов из-за высокого уровня ЛПНП происходит помутнение роговицы, появляются ксантомы на локтях и коленах, на подошве. При очень высоком уровне триглицеридов (превышающим 11,3 ммоль/л) развивается панкреатит и на теле наблюдаются ксантоматозные высыпания.
Мукополисахаридозы
Группа наследственных заболеваний соединительной ткани, характеризующихся сочетанным поражением опорно-двигательного аппарата, внутренних органов, глаз и нервной системы. Увеличены экскреция мукополисахаридов с мочой и накопление их в разных органах и тканях. Различают девять типов болезни. Вот некоторые из них.
47. Синдром МПСИ. Страдает фермент идуронидаза. Выражена умственная ретардация. Возможен пренатальный диагноз. Выявляются разные соматические аномалии. Тип наследственной передачи аутосомный рецессивный.
Болезням 48–52 свойственны сходные соматические нарушения: различная степень поражения костей, гепатоспленомегалия, ограничения подвижности суставов и др.
48. Болезнь Гунтера II. Страдает фермент идуронат сульфаза. Выражена умственная ретардация. Наблюдаются соматические аномалии. Возможен пренатальный диагноз. Тип наследственной передачи рецессивный, сцепленный с хромосомой X.
49. Синдром Санфилиппо III. Умственная отсталость сочетается с такими аномалиями, как утолщение костей черепа, увеличение печени, деформация поясничных позвонков. Возможен пренатальный диагноз. Тип наследственной передачи аутосомный рецессивный. Страдают разные ферменты (фосфатазы типы А-Д).
50. Болезнь Моркио IV. Страдает фермент N-ацетилгалактозамин. Данные об умственной ретардации отсутствуют. Возможен пренатальный диагноз. Тип наследования аутосомный рецессивный.
51. Болезнь Марото-Лами IV. Страдает фермент арилсульфатаза В. Умственная отсталость бывает не всегда. Возможен пренатальный диагноз. Тип наследования аутосомный рецессивный.
52. Гаргоилизм (название от изображений химер, украшавших старинные водосточные трубы). Другие названия: болезнь Гурлер, болезнь Джонни Мак-Л. (имя первого больного), множественный дазостоз, болезнь Пфаундлера. Основными симптомами являются: когнитивный дефицит, хондроостеодистрофия, гепатоспленомегалия, помутнение роговицы. Типичны большая голова, уродливое строение лицевой части черепа, карликовый рост, нарастающая слепота. Тип наследственной передачи аутосомный рецессивный. Возможна пренатальная диагностика болезни.
Классификация нарушения липидного обмена
На сегодняшний день основной считается классификация дислипидемий по Фредриксону:
- Причиной развития дислипидемии 1 типа является ферментативная недостаточность. Нарушения такого типа встречаются достаточно редко.
- Дислипидемия 2а возникает из-за мутаций в генах и относится к наиболее часто встречаемым типам нарушения липидного обмена.
- Дислипидемия 2б также встречается довольно часто и бывает, как наследственная, так и комбинированная (причиной могут быть перенесенные заболевания и питание, перенасыщенное животными жирами).
- Для нарушения липидного обмена по 3 типу характерно повышение в крови триглицеридов и ЛПНП.
- Эндогенное происхождение имеет гиперлипидемия 4 типа, характеризующаяся повышением ЛПОНП.
- Дислипидемия 5 типа возникает при увеличении в крови уровня холиномикронов и тоже относится к наследственным нарушениям.
Наследственные болезни обмена липидов
Наследственные болезни обмена липидов — обширная группа заболеваний, возникающих в результате нарушения одной из стадий синтеза, транспорта и деградации липопротеинов, в состав которых входят все основные плазменные липиды — триглицериды (ТГ), фосфолипиды, холестерол (ХС) и свободные жирные кислоты. Липиды плазмы крови наиболее часто находятся в соединении с различными апопротеинами, образуя липопротеины. К настоящему времени лучше всего изучены 8 основных апопротеинов — Аро-А 1,2,4, Аро-В, Аро-С 1,2,3 и Аро-Е. Некоторые апопротеины являются составной частью определенных липопротеинов, другие же могут мигрировать от одного липопротеина к другому.
Существует четыре основных класса липопротеинов плазмы крови:
1) хиломикроны (ХМ) — образуются в клетках слизистой оболочки кишечника и являются носителями пищевых ТГ;
2) липопротеины очень низкой плотности (ЛПОНП) — образуются в печени и используются для экспорта ТГ в другие ткани;
3) липопротеины низкой плотности (ЛПНП) — являются конечными продуктами катаболизма ЛПОНП и переносчиками ХС в клетки;
4) липопротеины высокой плотности (ЛПВП) — осуществляют метаболизм ХМ и ЛПОНП и транспортируют ХС из клеточных мембран в печень.
Типичный липопротеин состоит из липидного ядра, образованного триглицеридами и эфирами холестерола и наружного слоя, представленного фосфолипидами, холестеролом и апопротеинами.
Нормальный метаболизм липидов происходит следующим образом. Синтезированные в кишечнике ХМ и в печени ЛПОНП попадают в кровеносное русло и переносят ТГ (триглицериды) в жировые клетки и другие ткани. После отделения ТГ в жировых клетках, ХМ вновь поступают в печень, а ЛПОНП превращаются в ЛПНП, обогащенные ХС и содержащие Аро-В и Аро-Е. ЛПНП сохраняются в кровеносном русле в течение 2,5 суток и служат транспортерами ХС в различные клетки. Поступление ХС в клетки осуществляется при помощи рецепторов ЛПНП, количество и функции которых находятся под генетическим контролем. Клеточные рецепторы группируются в специальных участках клеточной мембраны, погруженных виде кратера в цитоплазму. После взаимодействия рецептора с ЛПНП участок мембраны, содержащий рецепторы, втягивается внутрь клетки и образует окаймленную везикулу. В цитоплазме ЛПНП отделяются от рецептора и проникают в лизосомы, а рецепторы возвращаются на клеточную мембрану. Освобождение ХС из ЛПНП осуществляется под действием нескольких ферментов. Обратная доставка ХС в клетки печени и тонкого кишечника осуществляется ЛПВП также с использованием механизма рецепторного эндоцитоза.
К настоящему времени описано более 420 различных мутаций, которые в зависимости от воздействия можно сгруппировать в 5 классов:
1) приводящие к прекращению синтеза рецептора;
2) нарушающие миграцию рецептора из эндоплазматического ретикулума в аппарат Гольджи;
3) изменяющие процесс связывания рецептора с ЛПНП;
4) нарушающие кластеризацию рецептора на клеточной мембран;
5) нарушающие процесс отщепления ЛПНП в клетке и транспорт рецептора к мембране клетки.
Основные этапы обмена ХС в организме находятся под сложным генетическим контролем. Нарушение в результате мутаций различных этапов этого метаболического пути может привести к задержке выведения ХС из организма и возникновению различных вариантов гиперлипидемий, развитию атеросклероза сосудов сердца и мозга. В последние годы удалось расшифровать молекулярно-генетические механизмы некоторых моногенных болезней, обусловленных нарушением обмена липидов.
Липоидозы — большая группа наследственных аномалий липидного обмена, общей чертой которых является высокий уровень липидов в плазме крови (плазматические липоидозы) либо накопление метаболитов липидного обмена внутри клеток (внутриклеточные липоидозы). К липоидозам относят болезни обмена липопротеидов. Наиболее распространенными формами липоидозов являются болезни Гоше и Ниманна-Пика, Тей-Сакса, относящиеся к так называемым «болезням накопления», клиническая дифференцировка которых крайне трудна.
Болезнь Гоше относится к сфинголипидозам — болезням накопления липидов; обусловлена дефектом гена, ответственного за синтез лизосомального гидролитического фермента бета — глюкоцереброзидазы (бета — гликозидазы).
Заболевание наследуется по аутосомно-рецессивному типу.
Дефект и дефицит этого фермента ведут к нарушению утилизации липидов — глюкоцереброзидов и накоплению их в макрофагах преимущественно костного мозга, селезенки, печени. Диагноз устанавливают по обнаружению специфических клеток Гоше (лимфоцитоподобное ядро, эксцентрично расположенное, и очень широкая светлая цитоплазма с чуть приметной циркулярной исчерченностью) в пунктате селезенки (пункцию ее можно делать только в стационаре) или в костном мозге.
Выделяют три типа болезни Гоше. Тип 1 (доброкачественный) – взрослый тип. В 30 раз чаще встречается у евреев (западноевропейской группы Ашкенази). Неврологические нарушения при этом отсутствуют, висцеральные изменения связаны преимущественно с кроветворными органами, увеличением селезенки, явлениями гиперспленизма, деструкцией костной ткани.
Тип 2 – инфантильный. Представляет собой злокачественную форму процесса с грубыми неврологическими нарушениями, которые проявляются уже у новорожденных и ведут к смерти в первые 2 года жизни.
Тип 3 – ювенильный. Отличается вариабельностью висцеральных и неврологических нарушений; по течению он менее злокачествен, чем тип 2. Разнообразие форм болезни Гоше обусловлено гетерогенностью мутаций гена бета — гликозидазы.
Бозень Нимана — Пика (сфингомиелиноз). Относится к группе тезауризмозов (болезней накопления) и характеризуется тяжелым поражением нервной системы, задержкой общего развития ребенка.
Наследование болезни происходит по аутосомно-рецессивному типу. Мальчики и девочки болеют одинаково часто. Нередко отмечается кровное родство между родителями, имеются семейные случаи болезни Нимана — Пика.
Патогенез болезни связывается с нарушением синтеза сфингомиелина, в составе которого отмечается избыточное количество одних жирных кислот и недостаток других. Возможно, определенное значение имеют нарушения процессов распада сфингомиелина, что приводит к его избыточному накоплению в ретикуло-эндотелиальных клетках различных органов и тканей. Микроскопически в селезенке, печени, почках, надпочечниках, лимфатических узлах, костном мозге и некоторых других органах обнаруживаются клетки Пика. Это довольно крупные клетки, имеющие размер от 20 до 50 мк и содержащие одно или много ядер. Протоплазма клеток содержит вакуоли, придающие клеткам характерный пенистый вид. Пенистость клеток образуется вследствие растворения липидных субстанций при фиксировании препарата. Гистологические изменения обнаруживаются в ганглиозных клетках головного мозга и сетчатой оболочки глаза. Количество сфингомиелина в различных органах резко увеличивается. В 1961 году, была предложена следующая классификация заболевания:
— болезнь Ниманна-Пика, тип А: классическая инфантильная;
— болезнь Ниманна-Пика, тип В: висцеральная;
— болезнь Ниманна-Пика, тип С: неострая подростковая
— болезнь Ниманна-Пика, тип D: ново-шотландская;
Однако, на сегодня, когда понятна генетическая природа заболевания, расстройство классифицируется следующим образом:
— болезнь Ниманна-Пика, связанная с геном SMPD1, которая включает в себя типы А и В;
— болезнь Ниманна-Пика, типа C, который включает в себя типы C1 и C2. (Тип D возникает в результате мутации того же гена, что и тип C1).
Клинически болезнь Нимана — Пика проявляется обычно в грудном возрасте, чаще в первое полугодие. Имеются единичные описания возникновения клинических признаков болезни в более старшем детском и даже молодом возрасте. Начальными симптомами болезни являются потеря аппетита, резкое похудание ребенка, задержка психофизического развития. Живот больного значительно увеличивается вследствие гепатоспленомегалии (увеличение селезенки и печени). Часто возникают бронхопневмонии. Лимфатические железы в большинстве случаев увеличиваются. Кожные покровы выглядят восковидными, блестящими, обнаруживаются участки пигментации. Неврологически в начальных стадиях болезни выявляются двигательные расстройства центрального характера: парезы конечностей, повышение тонуса мышц и сухожильных рефлексов, пирамидные знаки. В более поздних стадиях характерна гипотония мышц, отсутствие сухожильных рефлексов. Развивается идиотия, слепота и глухота. На глазном дне у многих больных могут обнаруживаться атрофия сосков зрительного нерва, вишнево-красное пятнышко овальной формы в макулярной области. Болезнь Нимана — Пика обладает злокачественным течением. Большинство детей погибают в первые 2 года жизни от легочно-сердечной недостаточности, интеркуррентных инфекций.
Болезнь Тея-Сакса — наследственное нейрометаболическое заболевание, связанное с накоплением в головном мозге ганглиозида GM2. Ганглиозиды содержат один или более остатков сиаловой кислоты.
Аутосомно-рецессивный тип наследования. Болезнь Тея-Сакса обусловлена мутационными поражениями гена гексозаминидазы (HEXA), контролирующего синтез альфа субъединицы гексозаминидазы А. Гексозаминидаза А — лизосомный фермент, катализирующий катаболизм GM2 ганглиозида.
Различают три формы болезни Тея — Сакса:
Детская форма — через полгода после рождения у детей отмечается прогрессирующее ухудшение физических возможностей и умственных способностей: наблюдаются слепота, глухота, потеря способности глотать. В результате атрофии мышц развивается паралич. Смерть наступает в возрасте до 3—4 лет.
Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания) дизартрия, (расстройства речи), атаксия (шаткость походки), спастичность (контрактуры и параличи). Смерть наступает в возрасте до 15—16 лет.
Взрослая форма — возникает в возрасте от 25 до 30 лет. Характеризуется симптомами прогрессирующего ухудшения неврологических функций: нарушение и шаткость походки, расстройства глотания и речи, снижение когнитивных навыков, спастичность, развитие шизофрении в форме психоза.
Болезнь Фабри является редкой наследственной Х-сцепленной рецессивной лизосомальной болезнью накопления. Дефицит фермента альфа-галактозидазы А, который возникает из-за мутации, вызывает накопление гликолипида известного как глоботриаосилцерамид (сокращенно GB3, GL-3) в кровеносных сосудах, других тканях и органах. Такое накопление приводит к нарушению их нормального функционирования.
Боль во всем теле или локализованные боли в конечностях (известный как акропарестезия) или желудочно-кишечном тракте (ЖКТ) является характерным явлением для пациентов с болезнью Фабри. Считается, что акропарестезия при заболевании связана с повреждением периферических нервных волокон, передающих боль. Боль в желудочно-кишечном тракте, вероятно, обусловлена накоплением липидов в малых его сосудах, что в свою очередь, мешает кровообращению (ишемия кишечника), вызывая боль. Распространенным и серьезным следствием болезни являются осложнения, связанные с почками. Почечная недостаточность (уремия) может ухудшаться со временем. Протеинурия (которая может вызвать пенистость мочи) часто является первым признаком поражения почек. Конечная стадия почечной недостаточности у мужчин, как правило, наступает в 30-40 летнем возрасте, и является частой причиной смерти. Ангиокератомы (маленькие, безболезненные папулы, которые могут появляться на любой части тела, но обычно появляются на бедрах, вокруг пупка, ягодицах, нижней части живота и в паху) тоже является распространенным симптомом. Ангидроз (отсутствие потоотделения) является распространенным симптомом в отличие от гипергидроза (повышенная потливость), который встречается значительно реже. При заболевании характерны также поражение глаз, а именно помутнение роговицы (известное как кератопатия).
Наследственный дефицит печеночной липазы,или гипер-a-триглицеридемия, характеризуется накоплением ТГ во фракции a-ЛП. В результате торможения катализируемых печеночной липазой реакций в крови. Проявления: липоидная дуга роговицы, ксантомы, ладонные стрии, ИБС.
Гиперлипидемия — повышение уровня липидов в крови или тканях, обусловленное врожденным или приобретенными нарушением обмена веществ.
Генетические факторы взаимодействуют с факторами окружающей среды, включающими образ питания и прием лекарств. Наследственные компоненты часто являются полигенными, и плохо поддаются определению, но тем не менее описано три чисто наследственных нарушения — семейная гиперхолестеринемия (СГХК), семейная гиперлипопротеинемия III типа и семейная комбинированная гиперлипидемия.
Различают пять типов гиперлипопротеинемий с характерными для каждого типа специфическими симптомами:
Тип I характеризуется приступами боли в желудке, обычно после употребления жирной пищи, а также общим ухудшением самочувствия, потерей аппетита и повышением температуры.
Тип II характеризуется появлением плотных образований на ахилловых сухожилиях и сухожилиях кистей рук и стоп
Тип III может вызывать появление над локтями и коленями мягких воспаленных язвочек. Тип IV обусловлен перееданием, ожирением и диабетом.
Тип V проявляется болями в животе (самый распространенный симптом), желтыми узелками на коже и красновато-беловатыми сосудиками на сетчатке глаз
Наследственная гиперхолестеринемия – аутосомно-доминантное, моногенное заболевание, причиной развития которого является нарушение структуры или функции рецепторов к липопротеинам низкой плотности (ЛПНП) на соматических клетках (печёночных и других) и/или их количества [5,13] (классическая форма НГХС [5]), или нарушения в структуре молекулы аполипопротеинов В-100 и С (апо В-100 и С, белковых компонентов липопротеинов).
В зависимости от природы генетического дефекта это заболевание классифицируется как рецептор-негативная, рецептор-дефективная или рецептор-интернационализационная формы.
Основными нарушениями являются нарушенный катаболизм ЛПНП печенью и другими тканями через ЛПНП-рецепторы, которые связываются с Apo В. При патологии ЛПНП-рецепторов это вызывает повышение уровня ЛПНП в крови и увеличение времени циркуляции данного вида липопротеинов, что в конце концов ведёт к образованию изменённых, модифицированных форм ЛПНП, обладающих высокой атерогенностью. Гомозиготные формы НГХС отличаются от гетерозиготных полным отсутствием ЛПНП-рецепторов на поверхности клеток и удаления ЛПНП из кровотока данным путём, и более длительной циркуляцией их периферическом кровяном русле в результате данных изменений (захват ЛПНП клетками происходит нерецепторным способом), тогда как при гетерозиготных формах число рецепторов снижено примерно на половину.
На коже больных НГХС отмечаются патогноматические признаки нарушения липидного обмена: ксантомы и ксантелазмы (отложение эфиров ХС в толще кожи).
Диагностические мероприятия
Диагностические процедуры включают консультацию врача, физикальный осмотр, сбор анамнеза и биохимический анализ крови.
При физикальном осмотре врач отмечает возможное наличие ксантелазмы, ксантом, липоидной дуги роговицы. Нередко у пациентов наблюдается повышенное артериальное давление. Аускультация (выслушивание) и перкуссия (простукивание) не являются информативными, так как не сопровождаются изменениями при дислипидемии.
Для выявления воспаления и сопутствующих заболеваний назначается лабораторный анализ крови и мочи. Генетический анализ выявляет гены — носители наследственной информации, отвечающие за развитие дислипидемии 2 типа.
Биохимический анализ крови позволяет определить уровень общего белка крови и сахара, мочевой кислоты и креатинина для обнаружения сопутствующего поражения органов. Иммунологический анализ определяет содержание антител к цитомегаловирусу и хламидиям, а также уровень С-реактивного протеина.
Но основным методом диагностики дислипидемии является липидограмма – анализ крови на жироподобные вещества, липиды. При измерении липидного спектра
определяются уровни триглицеридов, общего холестерина, ЛПНП и ЛПВП.
Исследования проводятся натощак, в собственной лаборатории НИАРМЕДИК.
Лечение дислипидемии назначается после полной диагностики, точной постановки диагноза и определения причины патологии.
Олигосахаридный и глюкопротеидный дисметаболизм
53. Болезнь клеточных включений. Страдают ферменты гликопротеид, N-ацетилглюкозаминилфосфотрансфераза. Выраженная умственная отсталость, а в соматическом плане — гепатомегалия, нарушения костной системы, опухание десен. Возможен пренатальный диагноз. Тип наследования аутосомный рецессивный.
54. Маннозидоз. Страдает фермент маннозидаза. Выраженная умственная отсталость. Соматически: гепатомегалия, нарушения костной системы, огрубение лица. Возможен пренатальный диагноз. Тип наследования аутосомный рецессивный.
55. Фукозидоз. Страдает фермент фукозидаза. Выраженная умственная ретардация. Соматически (см. п. 56). Возможен пренатальный диагноз. Тип наследования аутосомный рецессивный.
Лечение дислипидемии
Лечение дислипидемии комплексное и включает:
- Медикаментозную терапию – фибраты, витамины, статины и прочие препараты, корректирующие нарушения жирового обмена;
- Немедикаментозное лечение – нормализация веса путем дробного питания, дозированные физические нагрузки, ограничение алкоголя и курения, стрессовых ситуаций.
- Диетотерапию – рекомендуются продукты, богатые пищевыми волокнами и витаминами (овощи, злаковые, фрукты, бобы, обезжиренные молочнокислые продукты), не разрешается употреблять жирное и жареное мясо.
Если нарушение липидного баланса является вторичной патологией, возникшей вследствие воздействия негативных факторов или какого-либо заболевания, кардиологи НИАРМЕДИК назначают терапию, направленную на своевременное выявление и лечение основного заболевания.
Дислипидемия развивается годами и требует такого же длительного лечения. Предотвратить дальнейшее нарушение обмена липидов можно, точно соблюдая рекомендации врачей: больше двигаться, следить за весом, бросить вредные привычки.
Обращайтесь к врачам вовремя!
На ранних этапах остановить патологический процесс значительно проще. Своевременная терапия, устранение факторов риска и дисциплинированное выполнение рекомендаций врачей значительно продлевают и улучшают жизнь пациентов
Обращайтесь в наши клиники, не откладывайте визит к врачу. Вас проконсультируют опытные врачи, вы пройдете экспертное обследование на высокотехнологичной диагностической аппаратуре. На основании полученных результатов кардиолог назначит компетентную терапию заболевания дислипидемии и порекомендует профилактические мероприятия.
Чтобы записаться на прием к кардиологу, позвоните по телефону или оформите заявку на сайте.
Наши клиники в Санкт-Петербурге
Структурное подразделение Поликарпова Аллея Поликарпова 6к2 Приморский район
- Пионерская
- Удельная
- Комендантский
Структурное подразделение Жукова Пр.Маршала Жукова 28к2 Кировский район
- Автово
- Проспект Ветеранов
- Ленинский проспект
Структурное подразделение Девяткино Охтинская аллея 18 Всеволожский район
- Девяткино
- Гражданский проспект
- Академическая
Получить подробную информацию и записаться на прием Вы можете по телефону +7 (812) 640-55-25
Записаться на прием
Выделяют 5 видов липопротеидов:
- Хиломикроны синтезируются клетками кишечника, после чего попадают в лимфатические сосуды и далее в кровь. Являются транспортной формой триглециридов, холестерина и экзогенных жировых кислот.
- Липопротеиды очень низкой плотности. Они образуются в печени и содержат в себе до 15-ти процентов общего холестерина. Липопротеиды очень низкой плотности способны трансформироваться в мелкие частицы, большая часть которых выводится с печенью.
- Липопротеиды промежуточной плотности представляют собой промежуточную фазу липопротеидов очень низкой плотности.
- Липопротеиды низкой плотности (ЛНП) содержат в себе 60-70% холестерина и считаются основными переносчиками его тканям. Роль липопротеидов низкой плотности наиболее изучена в процессе атерогенеза. При нём образуются более мелкие и плотные частицы. Данный вид нарушений часто встречается при сахарном диабете.
- Липопротеиды высокой плотности (ЛВП) в своём составе содержат около 20-30% холестерина. Играют очень важную роль при обратном транспорте холестерина из тканей в печень. Специалистами была установлена связь между снижением концентрации ЛВП в крови и риском возникновения ишемической болезни сердца.