Arrhythmogenic right ventricular dysplasia (ARVD), or arrhythmogenic right ventricular cardiomyopathy, is a progressive hereditary disease of the heart muscle. It is characterized by focal fibrofatty degeneration of the right ventricular myocardium within the so-called triangle of dysplasia, located between the outflow and inflow tracts of the right ventricle and the apex of the heart, clinically manifested by ventricular arrhythmias, including paroxysmal ventricular tachycardia (VVT) [1, 2]. However, at present, fatty infiltration of the pancreatic myocardium is not a pathognomonic sign, since it can occur in older people without the disease. Against the background of muscle tissue atrophy, the destruction of cardiomyocytes continues: the presence of residual cells is less than 60% (major criterion), with or without fibrous or fatty replacement [3]. ARVD is a rare disease, but is associated with a high risk of sudden cardiac death (SCD). A number of studies have shown a high infection rate of patients with cardiotropic viruses, such as enterovirus and parvovirus [3, 4].
The clinic was first described by the Italian physician Giovanni Maria Lancisi. ARVD has been described as a hereditary disease occurring across four generations in all members of the same family. Despite the fact that the disease is considered hereditary, the genetic cause of the development of the pathology occurs in less than half of the cases [4].
In 60% of patients with ARVD, the disease is caused by mutations in genes encoding desmosomal proteins. Currently, more than 100 genes and mutations are known that are involved in various forms of heart disease (hypertrophic, dilated and arrhythmogenic cardiomyopathy) [4, 5]. Numerous studies have shown that more than 10% of carriers of desmosomal protein gene mutations have a high risk of developing complex arrhythmias and SCD [6, 7].
Clinically, ARVD is characterized by ventricular tachycardia, cardiac conduction block, right ventricular dilatation, and heart failure.
At its onset, the disease can be asymptomatic for a long time, however, even in the absence of specific symptoms, the risk of life-threatening arrhythmias is high.
As ARVD progresses, a number of symptoms come to the fore: interruptions in cardiac function (>50% of cases), cardialgia (46% of cases), palpitations (60% of cases), syncope (40% of cases), SCD (26% of cases) [ 8,9]. In the clinical picture of the disease, signs such as increased fatigue, dizziness, shortness of breath, and decreased exercise tolerance may be present, indicating the presence or progression of heart failure (HF).
The development of ARVD has several stages (latent, electrical disturbances, right ventricular HF, biventricular HF).
During the latent stage, SCD may be the first and only manifestation, the occurrence of which can be provoked by active physical activity. As the stage of electrical disturbances develops, cardiac arrhythmias and morphological changes in the right ventricle are recorded. As the process progresses, diffuse myocardial damage develops, which can lead to the development of biventricular heart failure (HF), complicated by various cardiac arrhythmias (including atrial fibrillation). The final stage of the process is usually represented by the clinical picture of dilated cardiomyopathy [10].
In young people, ARVD is the second most common cause of sudden death after coronary heart disease [8].
Given the asymptomatic course of the disease or the development of SCD as the first manifestation, epidemiological data remain controversial. On average, the disease occurs with a frequency of 6 per 10 thousand inhabitants. According to other data, ARVD is one of the most common cardiomyopathies and occurs in at least 200 patients in a city with a population of 1 million people [8]. It is noted that the disease is more often diagnosed in men (about 80%) of middle age (up to 40 years) [5]. It is believed that approximately 17% of SCD cases occur in patients suffering from ARVD. Close attention began to be paid to the early diagnosis of the disease in people involved in professional sports after studying the causes of VVS in 16 young athletes E. Larsson et al. in 1999. The study revealed that ARVD was detected in every 4th patient. However, it is possible that in this case the frequency of occurrence of the disease is due to a more thorough examination of people in this category. The onset of tachycardia may be delayed for many years until the RV is significantly enlarged and the size of the arrhythmogenic substrate is not large enough to cause persistent ventricular tachyarrhythmia.
Given the possibility of a long latent course of ARVD, the diagnostic process remains difficult. In addition to standard examination methods, in patients with suspected ARVD, in some situations, magnetic resonance imaging (MRI), contrast cardiac ventriculography and endomyocardial biopsy (EMB) are indicated. To assess the structure of the myocardium, as well as electrical and functional changes in the pancreas, 3D anatomical mapping is performed [10].
The prognosis of the disease directly depends on the timely prevention of sudden cardiac death.
We present our own observation.
Patient V., 48 years old, an employee, was admitted to the cardiology department of the Road Hospital at the Nizhny Novgorod station with complaints of palpitations, which were accompanied by severe weakness, dizziness, and a feeling of lack of air. Since the age of 45, he has been experiencing interruptions in his heart function and has not sought medical help. Worsening for three months, when the above-described attacks of palpitations appeared. On the eve of hospitalization, a similar attack occurred while walking quickly, and for the first time there was a short-term loss of consciousness.
History of rare acute respiratory diseases. Served in the army. Physical activity - moderate, did not engage in sports. Doesn't smoke, drinks alcohol in moderation. My father died suddenly at the age of 47, the cause is unknown.
Inspection results. Correct physique, excess nutrition (body mass index - 29.6 kg/m2). The skin is clean and of normal color. On auscultation of the lungs, breathing is vesicular, there are no wheezes. Respiration rate - 16 per minute. Heart sounds are rhythmic, muffled, heart rate is 78 beats/min. Blood pressure - 115/85 mm Hg. The liver is not enlarged, there is no peripheral edema.
According to laboratory tests, clinical and biochemical tests without pathology, NT-proBNP - 125 pg/ml (upper limit of normal).
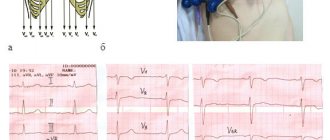
Data from instrumental examination methods. ECG: sinus rhythm, regular. Heart rate 76 beats/min. Horizontal position of the EOS. Violation of intraventricular conduction. T wave inversion in V1-V5. Epsilon wave in V1-V3 (Fig. 1).
Figure 1. Electrocardiogram of patient V.: intraventricular conduction disturbance, T wave inversion V1-V5, epsilon wave V1-V3.
Holter ECG monitoring: the study was carried out for 22 hours. During the day, sinus rhythm was recorded. The average heart rate is 77 beats/min, the minimum heart rate (37 beats/min) is during sleep, the maximum heart rate (168 beats/min) is when walking up the stairs. Ventricular ectopic activity is represented by 3412 monomorphic ventricular extrasystole (VES) with the morphology of left bundle branch block (LBBB), mainly in the daytime. Single - 3308, bigeminy type - 54, paired - 37 VES. Two episodes of unsustained ventricular tachycardia (VT) of 7 and 6 complexes were recorded (Fig. 2). No pauses, changes in ST-T, or QT interval were detected.
Figure 2. Holter monitoring of patient V.: paroxysm of unsustained VT with a frequency of 214 beats/min.
X-ray of the chest organs: no inflammatory or focal changes were detected. The pleural sinuses are free. The pulmonary pattern is somewhat enhanced. The roots of the lungs are not changed. The heart is positioned normally, the size is not enlarged. The vascular bundle is not changed. The aorta is without features. The superior vena cava is not dilated. The diaphragm is without any features.
According to transthoracic echocardiography (ECHO-CG, Fig. 3), an expansion of the cavity of the right ventricle (RV) was revealed with a decrease in its contractility (RV ejection fraction was 39%), an increase in the cavity of the right atrium to 6.1 × 4.6 cm and left atrium up to 5.6 × 5.0 cm. RV EDV - 164 ml; ESR of the pancreas - 102 ml. When examining the left ventricle (LV), no pathology was detected, LVEF was 51%. The inferior vena cava is not dilated, diameter is 2.0 cm, collapsed during inspiration by more than 50%. There was no evidence of congenital heart disease.
Figure 3. Echocardiogram of patient B. Dilatation of the atria and right ventricle.
Note: LA - left atrium; RA - right atrium; LV - left ventricle; RV - right ventricle; ESR - end-systolic size; EDV - end-diastolic size; ESV—end-systolic volume; EDV—end-diastolic volume; EF - ejection fraction.
Diagnostic coronary angiography: no data on hemodynamically significant stenoses of the coronary arteries were obtained.
To clarify the condition of the right side of the heart and exclude congenital defects, magnetic resonance imaging (MRI) of the heart was performed with gadolinium contrast enhancement.
Figure 4. Magnetic resonance imaging of patient B. Dyskinesis of the anterior wall of the right ventricle, microaneurysm of the anterior wall of the right ventricle, diastole phase.
Contrast-enhanced magnetic resonance imaging. The heart is positioned in a typical manner. RA 60 × 45 mm (enlarged), left atrium 48 × 50 mm (enlarged). Enlargement of the right ventricle: EDV - 166 ml, normalized EDV (EDV/body surface area (BSA)) - 111 ml/m2, RV ESV - 104 ml. RV ejection fraction - 39%. There is hypokinesis of the pancreatic walls with small areas of thinning and dyskinesis up to 3–4 mm (microaneurysms). Fatty infiltration of the pancreatic myocardium. LV: LV EDV - 45 mm, LV ESD - 34 mm, LV EDV - 155 ml, LV ESD - 68 ml, EF - 52%. The valves have not been changed. Conclusion: dilatation of the cavities of the right heart, decreased contractility of the right ventricle (EF - 39%); hypokinesis with areas of uneven thinning of the pancreatic walls; fatty infiltration of the pancreatic myocardium.
When performing programmed stimulation of the right ventricle, a paroxysm of monomorphic ventricular tachycardia (VT) was induced, the morphology of which was identical to the “jogs” of VT recorded during Holter ECG monitoring. The paroxysm stopped spontaneously after 4 seconds.
Based on the clinical picture, the results of laboratory and instrumental research methods, a diagnosis was made: Arrhythmogenic dysplasia of the right ventricle. Syncope from 04/13/2020
1.General information
Arrhythmogenic (causing arrhythmia) dysplasia/cardiomyopathy of the right ventricle (ARVD, ACM RV) - such a cumbersome name means a dangerous cardiac disease, known since the 18th century, but still insufficiently studied in a number of aspects.
The famous anatomist and materialist thinker of his time, the author of a number of outstanding works on malaria, plague of farm animals, syphilis, and the structure of the brain, the Italian Giovanni Maria Lancisi (1654-1720) was engaged in research, including the cardiovascular system. However, one of his main works, “De Motu Cordis et Aneurysmatibus”, which gave the first clinical description of arrhythmogenic pancreatic dysplasia, was published only posthumously, in 1728. Lancisi described a family disease that appeared in four generations and resulted unexpectedly and fatally (two and a half centuries later, this outcome would be called sudden cardiac death).
Modern nosological definitions of “arrhythmogenic dysplasia”, “arrhythmogenic right ventricular cardiomyopathy” appeared relatively recently, in the late 1970s - early 80s, and in ICD-10 they were classified under the heading “Other heart diseases / Cardiomyopathies”). The essence of this pathology repeats the patterns of many fibrosis, but also has pronounced distinctive features: the parenchymal muscle tissue of the heart, consisting of functional cardiomyocyte cells, degenerates and is replaced partly by fatty, partly by connective (scar) tissue, which inevitably affects the functioning of the myocardium as a whole, – causing, in particular, characteristic irregularities in heart rhythm, which gave the disease its name. The specificity is that the fibrosing process and fatty degeneration are almost always localized in the so-called. “dysplastic triangle” of the right ventricle of the heart muscle, i.e. in the area limited by the apex of the heart, the inlet and outlet tracts of the ventricle.
ACMP of the pancreas is considered a rare disease, but it has been established that it is the second most common cause of sudden cardiac death in young people. This is especially true for athletes, among whom such an outcome often becomes the first, only and last manifestation of the disease. In general, the proportion of ACMP of the pancreas as a cause of sudden cardiac death is, according to various estimates, from 20% to 25%, and in age samples under 20 years old this figure reaches 26%. About 80% of cases of arrhythmogenic right ventricular cardiomyopathy are diagnosed in people under the age of 40 years.
The incidence of ACMP of the pancreas in terms of the general population is estimated differently; Usually data is given at the level of 0.05% -0.5%. In the countries of the Mediterranean basin, the disease is registered much more often: up to 0.8% of the general population. The majority of patients are male.
However, all these data require careful verification and clarification in large samples, which is complicated by the complexity of diagnosis (in many cases, arrhythmogenic pancreatic dysplasia is not recognized and not registered as such) and the relative rarity of the disease.
A must read! Help with treatment and hospitalization!
Connective tissue dysplasia syndrome of the heart in children
In recent years, there has been an increase in the number of congenital malformations and hereditary diseases, as well as an increase in the prevalence of various variants of connective tissue dysplasia due to the deterioration of the environmental situation. According to modern concepts, connective tissue dysplasia syndrome is defined as an independent syndrome of a polygenic-multifactorial nature, manifested by external phenotypic signs in combination with dysplastic changes in connective tissue and clinically significant dysfunction of one or more internal organs (V. A. Gavrilova, 2002).
The term “cardiac connective tissue dysplasia” (CDTS) refers to an anomaly of tissue structure, which is based on a genetically determined defect in collagen synthesis. DSTS syndrome was identified as an independent nosological form at a symposium in Omsk (1990), dedicated to the problem of congenital connective tissue dysplasia. The problem of DSTS syndrome attracts attention due to the high risk of developing complications such as cardiac rhythm and conduction disturbances, infective endocarditis, thromboembolism of various vessels and sudden cardiac death.
The high frequency of DSTS syndrome in various diseases indicates the systemic nature of the lesion, which is associated with the “omnipresence” of connective tissue, which makes up the stroma of all organs and tissues.
Dysplastic heart is a combination of constitutional, topographical, anatomical and functional features of the heart in a person with connective tissue dysplasia (CTD). In Western literature, the term “myxoid heart disease” is used (Morales AB, Romanelli BEA, 1992), but this formulation is used mainly by foreign authors.
The incidence of dysplastic heart is 86% among individuals with primary undifferentiated DST (G. N. Vereshchagina, 2008).
According to modern concepts, DSTS syndrome includes prolapses of the heart valves, aneurysms of the interatrial septum and sinuses of Valsalva, ectopically attached chords of the mitral valve and many others.
The pathology is based on the inferiority of the extracellular matrix and its collagen structures.
A dysplastic heart is formed by:
I. Constitutional features - “drip”, “hanging” heart, its rotation around the sagittal and longitudinal axis.
II. Bone-vertebral dysplasia and deformities with compression, rotation, displacement of the heart and torsion of large vessels: according to Urmonas V.K. et al. (1983). Deformations of the chest and spine lead to the development of thoraco-phrenic syndrome, which limits the functioning of all organs of the chest.
III. Features of the structure of the heart and blood vessels:
- redundancy of tissue of the mitral, tricuspid and aortic valves;
- prolapse of the mitral valve leaflets (MVP) with regurgitation;
- myxomatous degeneration of leaflets, chords, valve ring;
- valvular-ventricular dissociation;
- bicuspid aortic valve;
- elongation, excessive mobility of the chordae;
- ectopically attached chordae;
- increased trabecularity of the left ventricle (LV);
- open oval window;
- aneurysm of the interatrial septum (small);
- dilatation of the sinuses of Valsalva;
- ventriculoseptal features of the LV: transient systolic ridge of the upper third of the interventricular septum (IVS), S-shaped bend of the IVS;
- tortuosity, hypoplasia, aplasia, fibromuscular dysplasia of the coronary arteries;
- coronary artery aneurysms;
- myocardial bridges;
- anomalies of the conduction system;
- expansion of the proximal part of the aorta, pulmonary trunk;
- aortic hypoplasia, borderline narrow aortic root, pulmonary trunk hypoplasia;
- systemic failure of the venous wall - varicose veins of the upper and lower extremities, pelvis, vulva, varicocele.
IV. Pathology of the respiratory system with a decrease in vital capacity of the lungs:
- diffuse and bullous emphysema;
- multiple fistulas;
- repeated spontaneous pneumothorax;
- bronchiectasis;
- cystic hypoplasia of the lungs.
Myxomatous degeneration of valves, chords, subvalvular structures is a genetically determined process of destruction and loss of the architectonics of collagen and elastic structures of connective tissue with the accumulation of acidic mucopolysaccharides in the loose fibrous layer. In this case, there are no signs of inflammation. It is based on a defect in the synthesis of type III collagen, which leads to thinning of the fibrous layer, the valves are enlarged, loose, redundant, the edges are curled, and sometimes fringe is detected. The primary locus of autosomal dominant myxomatosis in MVP is localized on chromosome 16. Morales AB (1992) identifies myxoid heart disease.
In population studies, the phenomenon of MVP was detected in 22.5% of children under the age of 12 years. In children with CTD, MVP is found much more often - in 45–68%.
Clinical manifestations of MVP in children vary from minimal to significant and are determined by the degree of connective tissue dysplasia of the heart, autonomic and neuropsychiatric abnormalities.
Most older children complain of short-term chest pain, palpitations, shortness of breath, a feeling of interruptions in the heart, dizziness, weakness, and headaches. Children characterize heart pain as stabbing, pressing, aching and feel it in the left half of the chest without any irradiation. They arise in connection with emotional stress and are usually accompanied by autonomic disorders: unstable mood, cold extremities, palpitations, sweating, and disappear spontaneously or after taking sedatives. The absence in most cases of ischemic changes in the myocardium according to a comprehensive examination allows us to regard cardialgia as a manifestation of sympathalgia associated with the psycho-emotional characteristics of children with MVP. Cardialgia with MVP may be associated with regional ischemia of the papillary muscles when they are excessively tense. Neurovegetative disorders are also associated with palpitations, a feeling of “interruptions” in the work of the heart, “tingling”, and “fading” of the heart. Headaches often occur during overwork, anxiety, in the morning before school starts and are combined with irritability, sleep disturbance, anxiety, and dizziness.
On auscultation, characteristic signs of mitral valve prolapse are isolated clicks (clicks), a combination of clicks with late systolic murmur, isolated late systolic murmur, holosystolic murmur.
The origin of the noise is associated with turbulent blood flow associated with bulging of the valves and vibration of the tense chords. Late systolic murmur is heard better in the left lateral decubitus position and intensifies during the Valsalva maneuver. The nature of the noise may change with deep breathing. As you exhale, the noise intensifies and sometimes takes on a musical tone. Often, the combination of systolic clicks and late murmur is most clearly detected in an upright position after exercise. Sometimes, when systolic clicks are combined with a late murmur in a vertical position, a holosystolic murmur may be recorded.
Holosystolic murmur with primary mitral valve prolapse is rare and indicates the presence of mitral regurgitation. This noise occupies the entire systole and practically does not change in intensity when changing body position, is carried out in the axillary region, and intensifies during the Valsalva maneuver.
The main methods for diagnosing MVP are two-dimensional Echo-CG and Dopplerography. MVP is diagnosed when the maximum systolic displacement of the mitral valve leaflets beyond the line of the mitral valve ring in the parasternal longitudinal position is 3 mm or more. The presence of isolated displacement of the anterior leaflet beyond the line of the mitral valve annulus in the four-chamber apical position is not enough to diagnose MVP; this is the main reason for its overdiagnosis.
Echo-CG classification of myxomatous degeneration (MD) (G. I. Storozhakov, 2004):
- MD 0 - no signs.
- MD I - minimally expressed: thickening of the leaflets 3–5 mm, arched deformation of the mitral orifice within 1–2 segments. The closure of the valves is preserved.
- MD II - moderately expressed: thickening of the leaflets 5–8 mm, elongation of the leaflets, deformation of the contour of the mitral orifice, its stretching, impaired closure of the leaflets. Mitral regurgitation.
- MD III - pronounced: thickening of the leaflets is more than 8 mm, the leaflets are elongated, multiple ruptures of the chords, significant expansion of the mitral ring, there is no closure of the leaflets. Multivalve lesion. Dilatation of the aortic root. Mitral regurgitation.
The degree of regurgitation in MVP depends on the presence and severity of myxomatous degeneration, the number of prolapsed leaflets and the depth of prolapse.
Degrees of regurgitation:
- 0—regurgitation is not recorded.
- I - minimal - the regurgitant jet penetrates into the cavity of the left atrium no more than one third of the atrium.
- II - medium - the regurgitation jet reaches the middle of the atrium.
- III - severe - regurgitation throughout the left atrium.
At rest, mitral regurgitation (MR) of the first degree is diagnosed in 16–20%, the second degree in 7–10% and the third degree in 3–5% of children with MVP.
The prognosis of a patient with MVP determines the degree of mitral regurgitation. Moreover, any degree of prolapse leads to changes in myocardial perfusion, changes most often in the area of the anterior wall of the LV and the interventricular septum (Nechaeva G.I., Viktorova I.A., 2007)).
Severe complications from MVP in children are rare. They are: life-threatening arrhythmias, infective endocarditis, thromboembolism, acute or chronic mitral regurgitation, and even sudden death.
Acute mitral regurgitation occurs due to the separation of the tendon threads from the cusps of the mitral valve (loppy mitral valve syndrome); it is rarely observed in childhood and is mainly associated with chest trauma in patients with myxomatous chordae degeneration. The main pathogenetic mechanism of acute mitral regurgitation is pulmonary venous hypertension, which occurs due to a large volume of regurgitation into an insufficiently distensible left atrium. Clinical symptoms are manifested by the sudden development of pulmonary edema.
In children, mitral regurgitation with MVP is most often asymptomatic and is diagnosed by Doppler echocardiography. Subsequently, as regurgitation progresses, complaints of shortness of breath during physical activity, decreased physical performance, weakness, and retarded physical development appear.
Risk factors for the development of “pure” (non-inflammatory) mitral regurgitation in prolapse syndrome according to two-dimensional echocardiography are:
- Dilatation of the left atrioventricular orifice.
- Prolapse of predominantly the posterior mitral leaflet.
- Thickening of the posterior mitral leaflet.
MVP is a high risk factor for infective endocarditis. The absolute risk of the disease is 4.4 times higher than in the population.
Diagnosis of infective endocarditis in MVP presents certain difficulties. Since the valves during prolapse are excessively scalloped, this does not allow us to detect the beginning of the formation of bacterial vegetations according to echocardiography. Therefore, the main importance in the diagnosis of endocarditis is played by: 1) clinical symptoms of the infectious process (fever, chills, rash, and other symptoms), 2) the appearance of the noise of mitral regurgitation and the fact of detection of the pathogen during repeated blood cultures.
The incidence of sudden death in MVP syndrome depends on many factors, the main ones being electrical instability of the myocardium in the presence of long QT interval syndrome, ventricular arrhythmias, concomitant mitral regurgitation, and neurohumoral imbalance.
The risk of sudden death in the absence of mitral regurgitation is low and does not exceed 2:10,000 per year, while with concomitant mitral regurgitation it increases 50–100 times.
In most cases, sudden death in patients with MVP is of arrhythmogenic origin and is caused by the sudden onset of idiopathic ventricular tachycardia (fibrillation) or against the background of long QT interval syndrome.
In rare cases, sudden cardiac death in patients with MVP may be due to a congenital anomaly of the coronary arteries (abnormal origin of the right or left coronary artery), leading to acute myocardial ischemia and necrosis.
Thus, the main risk factors for sudden death in children with MVP syndrome are: ventricular arrhythmias of grade III–V according to Lown; prolongation of the corrected QT interval more than 440 ms; the appearance of ischemic changes on the ECG during physical activity; history of cardiogenic syncope.
DSTS are one of the unfavorable factors predisposing to the development of arrhythmic complications in childhood and adolescence, including hemodynamically significant ones. In the structure of rhythm disturbances in children with DSTS, supraventricular extrasystole in pathological quantities and ventricular extrasystole, interrelated with the degree of cardiac dysplasia, are more often detected (Gnusaev S. F., co-authors, 2006).
Morphological manifestations of DSTS syndrome in children with concomitant kidney pathology, according to Domnitskaya T. M., Gavrilova V. A. (2000), are: spherical or triangular shape of the heart, rounding of the apex of the heart, an increase in heart weight by 1.4–2. 5 times, thickening and shortening of the mitral valve chords, fan-shaped discharge of the chords, hypertrophy of the papillary muscles, funnel-shaped mitral valve, open oval window. Myxomatous degeneration of the atrioventricular valve leaflets was observed in the majority of patients with DSTS syndrome and diseases of the urinary system (its frequency ranged from 66.7% to 77%). Endocardial fibroelastosis was detected in 10 children of the analyzed group.
In the population of children, the most frequently detected displacement of the septal leaflet of the tricuspid valve into the ventricular cavity within 10 mm, impaired distribution of chords of the anterior leaflet of the mitral valve, dilatation of the sinuses of Valsalva, enlarged eustachian valve more than 1 cm, dilatation of the pulmonary artery trunk, MVP, diagonally located trabeculae in the cavity left ventricle.
The management tactics for children with primary MVP vary depending on the severity of leaflet prolapse and the nature of autonomic and cardiovascular changes. The main principles of treatment are: 1) complexity; 2) duration; 3) taking into account the direction of functioning of the autonomic nervous system.
It is mandatory to normalize work, rest, daily routine, adherence to the correct regime with sufficient sleep.
The issue of physical education and sports is decided individually after the doctor evaluates the indicators of physical performance and adaptability to physical activity. Most children, in the absence of mitral regurgitation, severe disturbances in the repolarization process and ventricular arrhythmias, tolerate physical activity satisfactorily. If they have medical supervision, they can lead an active lifestyle without any restrictions on physical activity. Children can be recommended swimming, skiing, skating, and cycling. Sports activities associated with jerky movements (jumping, karate wrestling, etc.) are not recommended. The detection of mitral regurgitation, ventricular arrhythmias, changes in metabolic processes in the myocardium, and prolongation of the QT interval in a child dictates the need to limit physical activity and sports. These children are allowed to engage in physical therapy under the supervision of a doctor.
Treatment is based on the principle of restorative and vegetotropic therapy. The entire complex of therapeutic measures should be built taking into account the individual characteristics of the patient and the functional state of the autonomic nervous system.
An important part of the complex treatment of children with DSTS is non-drug therapy: psychotherapy, auto-training, physiotherapy (electrophoresis with magnesium, bromine in the upper cervical spine), water procedures, acupuncture, spinal massage. The doctor's attention should be directed to the sanitation of chronic foci of infection; tonsillectomy is performed according to indications.
Drug therapy should be aimed at: 1) treatment of vegetative-vascular dystonia; 2) prevention of the occurrence of myocardial neurodystrophy; 3) psychotherapy; 4) antibacterial prophylaxis of infective endocarditis.
For moderate manifestations of sympathicotonia, herbal medicine with sedative herbs, tincture of valerian, motherwort, herbal collection (sage, wild rosemary, St. John's wort, motherwort, valerian, hawthorn), which simultaneously has a slight dehydration effect, is prescribed. If there are changes in the repolarization process on the ECG, or rhythm disturbances, courses of treatment are carried out with drugs that improve metabolic processes in the myocardium (panangin, carnitine, Kudesan, vitamins). Carnitine is prescribed at a dose of 50 mg/kg per day for 2–3 months. Carnitine plays a central role in lipid and energy metabolism.
As a cofactor for beta-oxidation of fatty acids, it transports acyl compounds (fatty acids) across mitochondrial membranes, prevents the development of myocardial neurodystrophy, and improves its energy metabolism. In our studies, 35 children with extrasystole (more than 15 per minute) included carnitine in complex therapy. At the end of treatment, extrasystole decreased significantly in 25 children, and was not detected in 10 children.
A beneficial effect has been noted from the use of the drug Coenzyme Q10®, which significantly improves bioenergetic processes in the myocardium and is especially effective in secondary mitochondrial failure.
Early diagnosis of CTD in children allows for appropriate rehabilitation therapy and prevention of disease progression. One of the most striking therapeutic results is the effective treatment of children with DST (mainly with MVP) using the magnesium-containing drug magnesium orotate - Magnerot®. The choice of the drug was due to the known properties of the magnesium ion, observed in class I and IV antiarrhythmic drugs (membrane stabilizing and calcium antagonists), as well as the absence of side effects that may appear when using traditional antiarrhythmic therapy. It was also taken into account that the active ingredient of the drug is magnesium orotate, which, by inducing protein synthesis and participating in the exchange of phospholipids, which are an integral part of cell membranes, is necessary for the fixation of intracellular magnesium (O. A. Gromova, 2007).
The drug Magnerot® was used as monotherapy at a dose of 40 mg/kg per day during the first 7 days of administration, then 20 mg/kg per day for 6 months. The result of treatment was a decrease in the depth of prolapse of the mitral valve leaflets by 20–25% and a decrease in the degree of regurgitation by 15–17%. Treatment with Magnerot® did not affect the size of the left chambers of the heart and myocardial contractility, the values of which were within normal limits before treatment.
Studies conducted by E. N. Basargina (2008) revealed the antiarrhythmic effect of the drug Magnerot®. During daily ECG monitoring in children of groups 2 and 3, a decrease in the number of ventricular complexes by 50% or more was noted in 18 (27.7%) patients. Moreover, in 6 children, the disappearance of ventricular arrhythmia or a decrease in the number of ventricular complexes to 30–312 per day was noted. In 14 (21.5%) children, the number of ventricular complexes decreased by at least 30%. In two patients, an increase in the number of ventricular extrasystoles was noted up to 30% of the initial level. Thus, the antiarrhythmic effectiveness of Magnerot® was 27.7%. Similar results were previously obtained in other studies (Domnitskaya T. M. et al., 2005).
At the same time, rare supraventricular and ventricular extrasystoles, if not combined with long QT interval syndrome, as a rule, do not require the prescription of any antiarrhythmic drugs.
Thus, children with DSTS syndrome require timely diagnosis using Doppler echocardiography, electrocardiography, and in some cases 24-hour ECG monitoring, individual therapy, and monitoring by a pediatric cardiologist.
Therapy with Magnerot® in children with DSTS syndrome leads to a decrease in signs of valve prolapse, the frequency of detection of mitral regurgitation, a decrease in the severity of clinical manifestations of autonomic dysfunction, the frequency of ventricular arrhythmias, and is accompanied by an increase in the level of intraerythrocyte magnesium.
Literature
- Zemtsovsky E. V. Dysplastic syndromes and phenotypes. Dysplastic heart. St. Petersburg: "Olga". 2007. 80 p.
- Gavrilova V. A. Cardiac connective tissue dysplasia syndrome in children with diseases of the urinary system. Author's abstract. diss. Doctor of Medical Sciences M., 2002.
- Morales AB, Romanelli B., Boucek RJ et al. Myxoid heart disease: an assessment of extravalvular cardiac pathology in severe mitrae valve prolapse // Hum.Pathol. 1992, v. 23, no. 2, p. 129–137.
- Vereshchagina G. N. Systemic connective tissue dysplasia. Clinical syndromes, diagnosis, treatment approaches. Methodological manual for doctors. Novosibirsk, 2008, 37 p.
- Urmonas V.K., Kondrashin N.I. Funnel chest. Vilnius: Mokslas, 1983, 115 p.
- Gnusaev S. F. The significance of minor cardiac anomalies in healthy children and in cardiovascular pathology. Author's abstract. diss. Doctor of Medical Sciences, M., 1996.
- Belozerov Yu. M., Gnusaev S. F. Mitral valve prolapse in children. M.: Martis, 1995. 120 p.
- Storozhakov G.I., Vereshchagina G.S., Malysheva N.V. Assessment of individual prognosis for mitral valve prolapse // Cardiology, 2004, 4, p. 14–18.
- Nechaeva G. I., Viktorova I. A. Connective tissue dysplasia: terminology, diagnosis, patient management tactics. Omsk: Publishing house "Typography Blankom", 2007. 188 p.
- Gnusaev S. F., Belozerov Yu. M., Vinogradov A. F. Clinical significance of minor cardiac anomalies in children // Russian Bulletin of Perinatology and Pediatrics. 2006, no. 4. pp. 20–24.
- Domnitskaya T. M., Gavrilova V. A. Cardiac connective tissue dysplasia syndrome in children with diseases of the urinary system / Materials of the Second Congress of Pediatric Nephrologists of Russia. M., 2000. P. 159.
- Gromova O. A, Gogoleva I. V. The use of magnesium in the mirror of evidence-based medicine and fundamental research in therapy // Farmateka. 2007, v. 146, no. 12, p. 3–6.
- Basargina E. N. Cardiac connective tissue dysplasia syndrome in children // Issues of modern pediatrics. 2008, vol. 7, no. 1, 129–133.
- Domnitskaya T. M., Dyachenko A. V., Kupriyanova O. O., Domnitsky M. V. Clinical evaluation of the use of magnesium orotate in young people with cardiac connective tissue dysplasia syndrome // Cardiology. 2005; 45 (3): 76–81.
S. F. Gnusaev, Doctor of Medical Sciences, Professor
State Educational Institution of Higher Professional Education Tver State Medical Academy of Roszdrav , Tver
Contact information about the author for correspondence
2. Reasons
The genetic, hereditary nature of the disease applies to only 30-50% of cases, and the genes responsible for its development have already been identified. In the regional variant of ACMP of the pancreas, known as Naxos Island disease (Greece), fibrofatty degeneration and progressive heart failure are accompanied by congenital abnormalities of the skin and hair (keratoderma, “woolly hair”).
The etiopathogenesis of all other, non-hereditary cases remains essentially unknown. Hypotheses are considered and discussed according to which the degenerative process in the right ventricle of the myocardium is triggered as a result of bacterial or viral infectious inflammation; idiopathic (purely individual) features of intrauterine development; abnormally accelerated apoptosis (premature death of cardiomyocytes after a significantly lower number of cell divisions than normal); spontaneous degeneration of cardiomyocytes into cells of other types (transdifferentiation). However, even if any of these assumptions are confirmed statistically, it will be of little value without understanding the internal reasons: why arrhythmogenic dysplasia starts and develops in some people, while others do not.
Visit our Cardiology page
Symptoms of the appearance of arrhythmogenic dysplasia of the right ventricle
Sometimes the disease is asymptomatic for a long time, and its first manifestation may be sudden death.
In other cases, the patient is bothered by episodes of rapid heartbeat and fainting caused by paroxysmal ventricular tachycardia. With a less dangerous ventricular extrasystole, sometimes there is a feeling of interruptions in the heart.
Children and adolescents may complain of dizziness, unexpected attacks of palpitations or severe weakness, and fatigue.
As right ventricular failure progresses, swelling in the legs, heaviness in the right hypochondrium, abdominal enlargement, and weakness appear. If the function of the left ventricle is impaired, shortness of breath during physical exertion, attacks of suffocation in a lying position, and cough occur.
3. Symptoms and diagnosis
The clinical picture of ACMP of the pancreas requires differentiation from many other diseases and conditions in which gradually progressive heart failure is combined with episodes of arrhythmia. As a rule, patients begin to pay attention to increased heartbeat, interruptions in heart rhythm, shortness of breath, then pain in the heart, dizziness, sudden syncope with loss of consciousness (an unfavorable prognostic sign, which is considered one of the possible harbingers of sudden cardiac death). Fatigue increases, the patient tolerates physical activity less and less well.
However, in many cases, the progression of fibrofatty degeneration over a long period of time is not accompanied by any significant subjective sensations or clinical symptoms.
Considering the diagnostic complexity of ACMP of the pancreas, international cardiological associations have made attempts not only to unify terminology, but also to develop reliable recognition and differential diagnostic protocols. Thus, the presence of at least 3% connective tissue and at least 40% adipose tissue in the histological structure of the right ventricle is considered necessary to establish a diagnosis; Electrocardiographic, auscultatory and other diagnostic criteria are also used. For obvious reasons, a family history is required.
As necessary, EEG, echocardiography, stress tests, Holter monitoring, radiography, MRI are prescribed, and if all these methods do not allow an unambiguous diagnosis, it is necessary to resort to invasive endomyocardial biopsy, taking tissue samples for histological analysis.
About our clinic Chistye Prudy metro station Medintercom page!
What happens in the organ with Fontan's disease
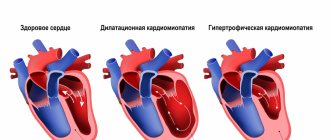
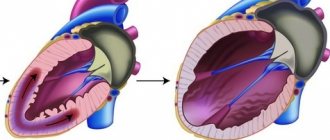
Structural abnormalities in this disease occur as a result of lipid impregnation and proliferation of connective tissue in the muscle of the right ventricle. This leads to its progressive expansion and decreased contractility. The left ventricle is affected much less frequently; the interventricular septum is practically not involved. Involvement of the left ventricle in the process is associated with a poor prognosis of the disease.
Mechanisms of destruction of myocardial cells:
- apoptosis (programmed cell death);
- inflammation, increasing fibrosis and loss of contractility;
- fatty replacement of heart cells.
Arrhythmogenic right ventricular dysplasia, autopsy of the heart of a young man who died suddenly while playing basketball.
As a result, the electrical stability of the myocardium is disrupted, and foci of pathological excitation appear in it. They become a source of ventricular arrhythmias.
Ventricular tachycardia that occurs suddenly can cause cardiac death in a young and apparently healthy person.
With further replacement of the myocardium with fatty and connective tissue, the function of the right ventricle suffers and its failure develops. At the final stages of the disease, there is already insufficiency of both ventricles, and the disease takes on the features of another cardiomyopathy - dilated.
4.Treatment
Etiopathogenetic therapy does not currently exist, and it will not appear until all issues related to the causes and pathogenesis of arrhythmogenic right ventricular dysplasia are finally clarified. The risk of sudden fibrillation and cardiac arrest is reduced by beta-blockers and other antiarrhythmic drugs; Cardiac glycosides, diuretics, ATP inhibitors, etc. are prescribed symptomatically. The effectiveness of such tactical schemes requires additional research. Lifestyle adjustments must be made, and any risk factors and provoking factors are strictly excluded. Some cardiac centers practice radiofrequency catheter ablation as a treatment, but this experience has not yet been studied enough.
There are opinions that the method of choice should be a radical intervention - a heart transplant or, at least, implantation of a cardioverter-defibrillator - however, to other specialists, the risk of possible complications during trans- or implantation seems justified only in cases where the process progresses quickly and manifests itself with obviously threatening symptoms.
Diagnosis of arrhythmogenic right ventricular dysplasia
The diagnosis is made when certain criteria are met. To detect them, the following studies are necessary:
- EchoCG;
- MRI of the heart;
- endocardial and myocardial biopsy;
- radioisotope angiography;
- ECG;
On the ECG of a patient with ARVD: negative T waves in leads V1–V5, arrows indicate epsilon waves.
- daily ECG monitoring;
- electrophysiological study of the heart;
- three-dimensional ECG mapping;
- molecular genetic testing.
Echocardiography for arrhythmogenic dysplasia of the right ventricle, see this video:
The “gold standard” for diagnosis is X-ray contrast ventriculography. During this test, a substance that is impenetrable to X-rays is injected into the heart cavity through a vein. Characteristic signs of the disease are:
- expansion (dilatation) of the right ventricle;
- impaired contractility of individual areas of the myocardium;
- protrusion in the area of affected cardiomyocytes;
- increased trabecularity - an increase in the number of muscle fibers in the cavity near the walls of the ventricle.
Many of these research methods are carried out only in specialized cardiology centers. Therefore, it is important to suspect the disease in time based on clinical data, and if unexplained dizziness, fainting, or palpitations occur, consult a doctor.
CRITERIA FOR DIAGNOSIS OF ARVD
Treatment of Fontan's disease
Considering the two main dangers of pathology - arrhythmia and heart failure, doctors use 2 areas of therapy:
- antiarrhythmic drugs are prescribed to prevent ventricular tachycardia: sotalol, amiodarone or flecainide;
- For heart failure, carvedilol and ACE inhibitors are used; for edema, diuretics and aldosterone antagonists are used.
If there is a significant slowdown in the heart rate, including that caused by taking antiarrhythmic drugs, a pacemaker may be installed. This device keeps the heart contracting when the heart rate slows down.
If drug therapy is ineffective, a cardioverter-defibrillator may be installed or radiofrequency ablation of the source of ventricular tachycardia may be performed. The latter option is used quite rarely, since its effectiveness in this particular disease is low.
A cardioverter-defibrillator is installed in patients with dilatation and decreased contractility of the right ventricle, involvement of the left ventricle, and the presence of ventricular tachycardia (according to EPI or HM ECG). This device is usually implanted in young people, and in almost 100% of cases it helps prevent sudden death caused by an arrhythmia attack.
In patients with severe rhythm disturbances combined with severe heart failure, ventriculotomy is performed. During the intervention, an anatomical interruption of the pathways along which the abnormal excitation of the heart occurs during arrhythmia occurs.
Heart transplantation is the most effective, but this operation is rarely performed.
I recommend reading about patent ductus arteriosus. You will learn about the etiology and hemodynamic disorders of PDA, symptoms of the disease, diagnostic measures and treatment. And here is more information about Wolff-Parkinson-White syndrome.
Etiology
Arrhythmogenic right ventricular cardiomyopathy is classified as rare because it is diagnosed in 1–6 people per 10 thousand of the population. The majority of patients are male; the disease occurs 4 times more often in men than in women.
Predisposing factors why arrhythmogenic cardiomyopathy develops remain unknown to specialists in the field of cardiology. However, clinicians have put forward several theories regarding the origin. The main one is genetic predisposition.
It is believed that the disease is transmitted in an autosomal dominant manner, and the mutant gene is located on chromosomes 12, 14, 17 and 18, which block specific proteins secreted by the myocardium. The prevalence of such a source of the disease ranges from 30 to 50%.
Among the theories of the origin of the disease, it is worth highlighting the following:
- Metabolic. The process of dysplasia may be associated with metabolic disorders occurring in the myocardium of the right ventricle. The basis for this assumption is a disease called muscular dystrophy of skeletal muscles with constant progression. However, it is unknown why dysplasia does not have a negative effect on the skeletal system.
- Inflammatory. It is based on the fact that in approximately 80% of patients with a similar diagnosis, inflammatory infiltration was detected during histological examination of the myocardium. The authors of the theory are confident that the disease is a consequence of myocarditis.
- Verified apoptosis (programmed cell death). Despite the existence of this assumption, it remains unknown why the degree of apoptosis is higher in the right ventricle and only in the absence of therapy spreads to the cardiac septum with subsequent damage to the left ventricle.
In isolated cases, the disease is inherited in an autosomal recessive manner - this variety is called Naxos disease. Currently, only 25 people in the world live with this pathology.