Аритмогенная дисплазия правого желудочка (АДПЖ), или аритмогенная кардиомиопатия правого желудочка, — прогрессирующее наследственное заболевание сердечной мышцы. Характеризуется очаговым фиброзно-жировым перерождением миокарда правого желудочка в пределах так называемого треугольника дисплазии, находящегося между выносящим, приносящим путём правого желудочка и верхушкой сердца, клинически проявляется желудочковыми нарушениями ритма, в том числе пароксизмальной желудочковой тахикардией (ЖТХ) [1, 2]. Однако в настоящее время жировая инфильтрация миокарда ПЖ не является патогномоничным признаком, поскольку она может встречаться у пожилых людей без заболевания. На фоне атрофии мышечной ткани продолжается разрушение кардиомиоцитов: наличие остаточных клеток — менее 60% (большой критерий), с фиброзным или жировым замещением или без него [3]. АДПЖ относится к редко встречающимся болезням, однако сопровождается высоким риском внезапной сердечной смерти (ВСС). В целом ряде исследований показана высокая инфицированность больных кардиотропными вирусами, такими как энтеровирус, парвовирус [3, 4].
Впервые клинику описал итальянский врачанатом Джованни-Мария Ланчизи. АДПЖ была описана как наследственное заболевание, наблюдающееся в четырех поколениях у всех членов одной семьи. Несмотря на то, что заболевание считается наследственным, генетическая причина развития патологии встречается менее чем в половине случаев [4].
У 60% пациентов с АДПЖ причиной заболевания являются мутации в генах, кодирующих десмосомные белки. В настоящее время известно более 100 генов и мутаций, принимающих участие в различных формах заболеваний сердца (гипертрофическая, дилатационная и аритмогенная кардиомиопатия) [4, 5]. В результате многочисленных исследований показано, что более 10% носителей мутаций генов десмосомных белков имеют высокий риск развития сложных аритмий и ВСС [6, 7].
Клинически АДПЖ характеризуется желудочковой тахикардией, блокадой проводящей системы сердца, дилатацией правого желудочка и сердечной недостаточностью.
В дебюте заболевание длительно может протекать бессимптомно, однако и в отсутствие специфической симптоматики высок риск жизнеугрожаемых аритмий.
При прогрессировании АДПЖ на первый план выходит ряд симптомов: перебои в работе сердца (>50% случаев), кардиалгии (46% случаев), сердцебиения (60% случаев), синкопальные состояния (40% случаев), ВСС (26% случаев) [8,9]. В клинике заболевания могут присутствовать такие признаки, как повышенная утомляемость, головокружение, одышка, снижение толерантности к физическим нагрузкам, указывающие на присутствие или прогрессирование сердечной недостаточности (СН).
Развитие АДПЖ имеет несколько стадий (латентную, электрических нарушений, правожелудочковой СН, бивентрикулярной СН).
Во время латентной стадии ВСC может быть первым и единственным проявлением, возникновение которой может провоцироваться активной физической нагрузкой. При развитии стадии электрических нарушений регистрируются нарушения ритма сердца и морфологические изменения правого желудочка. При прогрессировании процесса развивается диффузное поражение миокарда, которое может приводить к развитию бивентрикулярной сердечной недостаточности (СН), осложненной различными нарушениями ритма сердца (включая фибрилляцию предсердий). Финальная стадия процесса, как правило, представлена клиникой дилатационной кардиомиопатии [10].
В молодом возрасте АДПЖ является второй по частоте причиной внезапной смерти после ишемической болезни сердца [8].
Учитывая бессимптомное течение болезни или развитие ВСС как первого проявления, эпидемиологические данные остаются спорными. В среднем, заболевание встречается с частотой 6 на 10 тыс. жителей. По другим данным, АДПЖ является одной из самых распространенных кардиомиопатий и встречается, как минимум, у 200 пациентов в городе с населением 1 млн человек [8]. Отмечается, что болезнь чаще диагностируется у мужчин (около 80%) среднего возраста (до 40 лет) [5]. Считается, что у пациентов, страдающих АДПЖ, встречается примерно 17% случаев ВСС. Пристальное внимание стали уделять ранней диагностике заболевания у лиц, занимающихся спортом профессионально, после изучения причин ВВС у 16 молодых спортсменов E. Larsson и соавт. в 1999 г. В результате проведенного исследования выяснилось, что АДПЖ была обнаружена у каждого 4-го пациента. Однако возможно, что в данном случае частота встречаемости заболевания обусловлена более тщательным обследованием лиц данной категории. Начало тахикардии может быть отсрочено на многие годы, пока ПЖ значительно не увеличен, а размер аритмогенного субстрата недостаточно велик, чтобы вызвать постоянную желудочковую тахиаритмию.
Учитывая возможность длительного латентного течения АДПЖ, процесс диагностики остаётся сложным. Кроме стандартных методов обследования у пациентов с подозрением на АДПЖ в некоторых ситуациях показано проведение магниторезонансной томографии (МРТ), контрастной вентрикулографии сердца и эндомиокардиальной биопсии (ЭМБ). Для оценки структуры миокарда, а также электрических и функциональных изменений ПЖ проводится 3D-анатомическое картирование [10].
Прогноз заболевания напрямую зависит от своевременной профилактики внезапной сердечной смерти.
Представляем собственное наблюдение.
Пациент В., 48 лет, служащий, поступил в кардиологическое отделение Дорожной больницы на станции Нижний Новгород с жалобами на приступы сердцебиения, которые сопровождаются резкой слабостью, головокружением, чувством нехватки воздуха. С 45 лет отмечает перебои в работе сердца, за медицинской помощью не обращался. Ухудшение в течение трёх месяцев, когда появились вышеописанные приступы сердцебиения. Накануне госпитализации во время быстрой ходьбы возник подобный приступ, впервые была кратковременная потеря сознания.
В анамнезе редкие острые респираторные заболевания. Служил в армии. Физическая активность — умеренная, спортом не занимался. Не курит, алкоголь употребляет умеренно. Отец умер внезапно в 47 лет, причина неизвестна.
Результаты осмотра. Правильного телосложения, избыточного питания (индекс массы тела — 29,6 кг/м2). Кожные покровы чистые, обычной окраски. При аускультации легких дыхание везикулярное, хрипов нет. Частота дыхания — 16 в минуту. Тоны сердца ритмичные, приглушены, частота сердечных сокращений 78 уд/мин. Артериальное давление — 115/85 мм рт.ст. Печень не увеличена, периферических отёков нет.
По данным проведенных лабораторных исследований, клинические и биохимические анализы без патологии, NT-proВNP – 125 пг/мл (верхняя граница нормы).
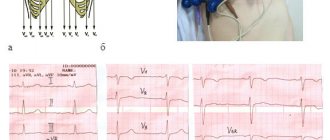
Данные инструментальных методов обследования. ЭКГ: ритм синусовый, регулярный. ЧСС 76 уд/мин. Горизонтальное положение ЭОС. Нарушение внутрижелудочковой проводимости. Инверсия зубца Т в V1-V5. Эпсилон волна в V1-V3 (рис.1).
Рисунок 1. Электрокардиограмма больного В.: нарушение внутрижелудочковой проводимости, инверсия зубца Т V1-V5, эпсилон-волна V1-V3.
Холтеровское ЭКГ мониторирование: исследование проводилось в течение 22 часов. За сутки зафиксирован синусовый ритм. Средняя ЧСС 77 уд/мин, ЧСС минимальная (37 уд/мин.) — во время сна, ЧСС максимальная (168 уд/мин) — при ходьбе по лестнице. Желудочковая эктопическая активность представлена 3412 мономорфной желудочковой экстрасистолией (ЖЭС) с морфологией блокады левой ножки пучка Гиса (БЛНПГ), преимущественно в дневное время. Одиночных — 3308, по типу бигеминии — 54, парных — 37 ЖЭС. Зарегистрированы два эпизода неустойчивой желудочковой тахикардии (ЖТ) по 7 и 6 комплексов (рис. 2.). Пауз, изменений ST-T, интервала QT не выявлено.
Рисунок 2. Холтеровское мониторирование больного В.: пароксизм неустойчивой ЖТ с частотой 214 уд/мин.
Рентгенография органов грудной клетки: воспалительных и очаговых изменений не выявлено. Плевральные синусы свободны. Легочный рисунок несколько усилен. Корни легких не изменены. Сердце расположено нормально, размеры не увеличены. Сосудистый пучок не изменен. Аорта без особенностей. Верхняя полая вена не расширена. Диафрагма без особенностей.
По данным трансторакальной эхокардиографии (ЭХО-КГ, рис. 3), выявлено расширение полости правого желудочка (ПЖ) со снижением его сократительной способности (фракция выброса ПЖ составила 39%), увеличение полости правого предсердия до 6,1 × 4,6 см и левого предсердия до 5,6 × 5,0 см. КДО ПЖ — 164 мл; КСО ПЖ — 102 мл. При изучении левого желудочка (ЛЖ) патологии выявлено не было, ФВ ЛЖ – 51%. Нижняя полая вена не расширена, диаметр — 2,0 см, коллабировала на вдохе более 50%. Данных о врождённом пороке сердца не выявлено.
Рисунок 3. Эхокардиограмма больного В. Расширение предсердий и правого желудочка.
Примечание: ЛП — левое предсердие; ПП — правое предсердие; ЛЖ — левый желудочек; ПЖ — правый желудочек; КСР — конечно-систолический размер; КДР — конечно-диастолический размер; КСО — конечно-систолический объем; КДО — конечно-диастолический объем; ФВ — фракция выброса.
Диагностическая коронарография: данных о гемодинамически значимых стенозах коронарных артерий не получено.
Для уточнения состояния правых отделов сердца и исключения врождённых пороков была проведена магнитно-резонансная томография (МРТ) сердца с контрастным усилением гадолинием.
Рисунок 4. Магнитно-резонансная томограмма больного В. Дискинез передней стенки правого желудочка, микроаневризмы передней стенки правого желудочка, фаза диастолы.
Магнитно-резонансная томография с контрастным усилением. Сердце расположено типично. ПП 60× 45 мм (увеличено), левое предсердие 48 × 50 мм (увеличено). Увеличение правого желудочка: КДО — 166 мл, нормализованный показатель КДО (КДО/ площади поверхности тела (ППТ)) — 111 мл/м2, КСО ПЖ — 104 мл. Фракция выброса ПЖ — 39%. Отмечается гипокинез стенок ПЖ с мелкими участками истончения и дискинеза до 3–4 мм (микроаневризмы). Жировая инфильтрация миокарда ПЖ. ЛЖ: КДР ЛЖ — 45 мм, КСР ЛЖ — 34 мм, КДО ЛЖ — 155 мл, КСО ЛЖ — 68 мл, ФВ — 52%. Клапаны не изменены. Заключение: дилатация полостей правых отделов сердца, снижение сократительной способности ПЖ (ФВ — 39%); гипокинез с участками неравномерного истончения стенок ПЖ; жировая инфильтрация миокарда ПЖ.
При выполнении программируемой стимуляции правого желудочка был индуцирован пароксизм мономорфной желудочковой тахикардии (ЖТ), морфология которого была идентична «пробежкам» ЖТ, зарегистрированным при холтеровском мониторировании ЭКГ. Пароксизм самостоятельно купировался через 4 сек.
На основании клинической картины, результатов лабораторных и инструментальных методов исследования поставлен диагноз: Аритмогенная дисплазия правого желудочка. Синкопальное состояние от 13.04.2020 г.
1.Общие сведения
Аритмогенная (вызывающая аритмию) дисплазия/кардиомиопатия правого желудочка (АДПЖ, АКМП ПЖ) – столь громоздким названием означается опасное кардиологическое заболевание, известное с ХVIII века, но до сих пор в ряде аспектов изученное недостаточно.
Прославленный врач-анатом и мыслитель-материалист своего времени, автор ряда выдающихся трудов о малярии, чуме сельскохозяйственных животных, сифилисе, строении мозга, – итальянец Джованни Мария Ланчизи (1654-1720) занимался исследованиями, в том числе, сердечнососудистой системы. Однако одна из главных его работ, – «De Motu Cordis et Aneurysmatibus», – в которой дано первое клиническое описание аритмогенной дисплазии ПЖ, была опубликована лишь посмертно, в 1728 году. Ланчизи описал семейную болезнь, появлявшуюся в четырех поколениях и результировавшую неожиданно и фатально (спустя два с половиной века такой исход станут называть внезапной сердечной смертью).
Современные нозологические определения «аритмогенная дисплазия», «аритмогенная кардиомиопатия правого желудочка» появились сравнительно недавно, в конце 1970 — начале 80-х годов, и в МКБ-10 они были отнесены к рубрике «Другие болезни сердца / Кардиомиопатии»). Суть данной патологии повторяет закономерности очень многих фиброзов, однако обладает и выраженными отличительными особенностями: паренхиматозная мышечная ткань сердца, состоящая из функциональных клеток-кардиомиоцитов, перерождается и вытесняется отчасти жировой, отчасти соединительной (рубцовой) тканями, что неизбежно сказывается на функционировании миокарда в целом, – вызывая, в частности, характерные сбои сердечного ритма, которые и дали название болезни. Специфика заключается в том, что фиброзирующий процесс и жировая дегенерация почти всегда локализованы в т.н. «диспластическом треугольнике» правого желудочка сердечной мышцы, т.е. в зоне, ограниченной верхушкой сердца, входными и выводными путями желудочка.
АКМП ПЖ считается редким заболеванием, однако же установлено, что оно выступает второй из наиболее распространенных причин внезапной сердечной смерти в молодом возрасте. В особенности это касается спортсменов, среди которых такой исход нередко становится первым, единственным и последним проявлением болезни. В целом, доля АКМП ПЖ как причины внезапной сердечной смерти составляет, по разным оценкам, от 20% до 25%, а в возрастных выборках младше 20 лет этот показатель достигает 26%. Около 80% случаев аритмогенной правожелудочковой кардиомиопатии диагностируется у лиц в возрасте до 40 лет.
Частота встречаемости АКМП ПЖ в пересчете на общую популяцию оценивается по-разному; обычно приводятся данные на уровне 0,05%-0,5%. В странах средиземноморского бассейна заболевание регистрируется существенно чаще: до 0,8% от общей популяции. Преобладают пациенты мужского пола.
Однако все эти данные требуют тщательной верификации и уточнения на больших выборках, что затрудняется сложностью диагностики (во многих случаях аритмогенная дисплазия ПЖ не распознается и не регистрируется как таковая) и относительной редкостью заболевания.
Обязательно для ознакомления! Помощь в лечении и госпитализации!
Синдром соединительнотканной дисплазии сердца у детей
В последние годы отмечается увеличение числа врожденных пороков развития и наследственных заболеваний, а также нарастание распространенности различных вариантов соединительнотканной дисплазии из-за ухудшения экологической обстановки. По современным представлениям синдром соединительнотканной дисплазии определяют как самостоятельный синдром полигенно-мультифакториальной природы, проявляющийся внешними фенотипическими признаками в сочетании с диспластическими изменениями соединительной ткани и клинически значимой дисфункцией одного или нескольких внутренних органов (В. А. Гаврилова, 2002).
Под термином «дисплазия соединительной ткани сердца» (ДСТС) подразумевается аномалия тканевой структуры, в основе которой лежит генетически детерминированный дефект синтеза коллагена. Синдром ДСТС был выделен в самостоятельную нозологическую форму на симпозиуме в г. Омске (1990), посвященном проблеме врожденной дисплазии соединительной ткани. Проблема синдрома ДСТС привлекает к себе внимание в связи с большим риском развития таких осложнений, как нарушения ритма и проводимости сердца, инфекционный эндокардит, тромбоэмболия различных сосудов и внезапная сердечная смерть.
Высокая частота синдрома ДСТС при различных заболеваниях свидетельствует о системности поражения, что связано с «вездесущностью» соединительной ткани, составляющей строму всех органов и тканей.
Диспластическое сердце — сочетание конституциональных, топографических, анатомических и функциональных особенностей сердца у человека с дисплазией соединительной ткани (ДСТ). В западной литературе используется термин «миксоидная болезнь сердца» (Morales A. B., Romanelli B. E. A., 1992), однако эта формулировка используется преимущественно зарубежными авторами.
Частота диспластического сердца составляет 86% среди лиц с первичной недифференцированной ДСТ (Г. Н. Верещагина, 2008).
По современным представлениям к синдрому ДСТС относят пролапсы клапанов сердца, аневризмы межпредсердной перегородки и синусов Вальсальвы, эктопически крепящиеся хорды митрального клапана и многие другие.
В основе патологии лежит неполноценность внеклеточного матрикса, его коллагеновых структур.
Диспластическое сердце формируют:
I. Конституциональные особенности — «капельное», «висячее» сердце, поворот его вокруг сагиттальной и продольной оси.
II. Костно-вертебральные дисплазии и деформации со сдавлением, ротацией, смещением сердца и перекрутом крупных сосудов: по данным Урмонаса В. К. и др. (1983). Деформации грудной клетки и позвоночника приводят к развитию торако-диафрагмального синдрома, ограничивающего работу всех органов грудной клетки.
III. Особенности строения сердца и сосудов:
- избыточность ткани створок митрального, трикуспидального и аортального клапанов;
- пролабирование створок митрального клапана (ПМК) с регургитацией;
- миксоматозная дегенерация створок, хорд, клапанного кольца;
- вальвулярно-вентрикулярная диссоциация;
- двустворчатый аортальный клапан;
- удлинение, избыточная подвижность хорд;
- эктопически крепящиеся хорды;
- повышенная трабекулярность левого желудочка (ЛЖ);
- открытое овальное окно;
- аневризма межпредсердной перегородки (небольшая);
- дилятация синусов Вальсальвы;
- вентрикуло-септальные особенности ЛЖ: транзиторный систолический валик верхней трети межжелудочковой перегородки (МЖП), S-образный изгиб МЖП;
- извитость, гипоплазия, аплазия, фибромускулярная дисплазия коронарных артерий;
- аневризмы коронарных артерий;
- миокардиальные мостики;
- аномалии проводящей системы;
- расширение проксимальной части аорты, легочного ствола;
- гипоплазия аорты, погранично узкий корень аорты, гипоплазия легочного ствола;
- системная несостоятельность венозной стенки — варикозное расширение вен верхних и нижних конечностей, малого таза, вульвы, варикоцеле.
IV. Патология органов дыхания со снижением жизненной емкости легких:
- диффузная и буллезная эмфизема;
- множественные свищи;
- повторные спонтанные пневмотораксы;
- бронхоэктазы;
- кистозная гипоплазия легких.
Миксоматозная дегенерация створок, хорд, подклапанных структур — генетически детерминированный процесс разрушения и утраты архитектоники коллагеновых и эластических структур соединительной ткани с накоплением в рыхлом фиброзном слое кислых мукополисахаридов. При этом признаки воспаления отсутствуют. В основе — дефект синтеза коллагена III типа, что приводит к истончению фиброзного слоя, створки увеличены, рыхлые, избыточные, края закручены, иногда определяется бахрома. Первичный локус аутосомно-доминантного миксоматоза при ПМК локализован в хромосоме 16. Morales A. B. (1992) выделяет миксоидную болезнь сердца.
В популяционных исследованиях феномен ПМК выявлен у 22,5% детей в возрасте до 12 лет. У детей с ДСТ ПМК обнаруживается значительно чаще — у 45–68%.
Клинические проявления ПМК у детей варьируют от минимальных до значительных и определяются степенью соединительнотканной дисплазии сердца, вегетативными и психоневрологическими отклонениями.
Большинство детей старшего возраста жалуются на кратковременные боли в грудной клетке, сердцебиение, одышку, ощущение перебоев в сердце, головокружение, слабость, головные боли. Боли в сердце дети характеризуют как колющие, давящие, ноющие и ощущают в левой половине грудной клетки без какой-либо иррадиации. Они возникают в связи с эмоциональным напряжением и сопровождаются, как правило, вегетативными нарушениями: неустойчивым настроением, похолоданием конечностей, сердцебиением, потливостью, проходят самопроизвольно или после приема седативных средств. Отсутствие в большинстве случаев ишемических изменений в миокарде по данным комплексного обследования позволяет расценить кардиалгии как проявление симпаталгии, связанной с психоэмоциональными особенностями детей с ПМК. Кардиалгии при ПМК могут быть связаны с региональной ишемией папиллярных мышц при их чрезмерном натяжении. С нейровегетативными нарушениями также связаны сердцебиение, ощущение «перебоев» в работе сердца, «покалывание», «замирание» сердца. Головные боли чаще возникают при переутомлении, переживаниях, в утренние часы перед началом занятий в школе и сочетаются с раздражительностью, нарушением сна, тревогой, головокружением.
При аускультации характерными признаками пролапса митрального клапана являются изолированные щелчки (клики), сочетание щелчков с позднесистолическим шумом, изолированный позднесистолический шум, голосистолический шум.
Происхождение шума связано с турбулентным током крови, связанным с выбуханием створок и вибрацией натянутых хорд. Позднесистолический шум выслушивается лучше в положении лежа на левом боку, усиливается при проведении пробы Вальсальвы. Характер шума может меняться при глубоком дыхании. На выдохе шум усиливается и иногда приобретает музыкальный оттенок. Нередко сочетание систолических щелчков и позднего шума наиболее отчетливо выявляется в вертикальном положении после физической нагрузки. Иногда при сочетании систолических щелчков с поздним шумом в вертикальном положении может регистрироваться голосистолический шум.
Голосистолический шум при первичном пролапсе митрального клапана наблюдается редко и свидетельствует о наличии митральной регургитации. Этот шум занимает всю систолу и практически не меняется по интенсивности при перемене положения тела, проводится в подмышечную область, усиливается при проведении пробы Вальсальвы.
Основными методами диагностики ПМК являются двухмерная Эхо-КГ и допплерография. ПМК диагностируют при максимальном систолическом смещении створок митрального клапана за линию кольца митрального клапана в парастернальной продольной позиции на 3 мм и более. Наличия изолированного смещения передней створки за линию кольца митрального клапана в четырехкамерной верхушечной позиции недостаточно для диагностики ПМК, это служит основной причиной его гипердиагностики.
Эхо-КГ-классификация миксоматозной дегенерации (МД) (Г. И. Сторожаков, 2004):
- МД 0 — признаков нет.
- МД I — минимально выраженная: утолщение створок 3–5 мм, аркообразная деформация митрального отверстия в пределах 1–2 сегментов. Смыкание створок сохранено.
- МД II — умеренно выраженная: утолщение створок 5–8 мм, удлинение створок, деформация контура митрального отверстия, его растяжение, нарушение смыкания створок. Митральная регургитация.
- МД III — резко выраженная: утолщение створок больше 8 мм, створки удлинены, множественные разрывы хорд, значительное расширение митрального кольца, смыкание створок отсутствует. Многоклапанное поражение. Дилятация корня аорты. Митральная регургитация.
Степень регургитации при ПМК зависит от наличия и выраженности миксоматозной дегенерации, количества пролабирующих створок и глубины пролабирования.
Степени регургитации:
- 0 — регургитация не регистрируется.
- I — минимальная — струя регургитации проникает в полость левого предсердия не более чем на одну треть предсердия.
- II — средняя — струя регургитации достигает середины предсердия.
- III — тяжелая — регургитация по всему левому предсердию.
В состоянии покоя митральная регургитация (МР) первой степени диагностируется у 16–20%, вторая степень — у 7–10% и третья степень — у 3–5% детей с ПМК.
Прогноз больного с ПМК определяет степень митральной регургитации. При этом любая степень пролабирования приводит к изменениям перфузии миокарда, изменениям чаще в области передней стенки ЛЖ и межжелудочковой перегородки (Нечаева Г. И., Викторова И. А., 2007)).
Тяжелые осложнения при ПМК у детей встречаются нечасто. Ими являются: жизнеугрожаемые аритмии, инфекционный эндокардит, тромбоэмболия, острая либо хроническая митральная недостаточность и даже внезапная смерть.
Острая митральная недостаточность возникает из-за отрыва сухожильных нитей от створок митрального клапана (синдром «болтающегося» клапана — loppy mitral valve), в детском возрасте наблюдается казуистически редко и в основном связана с травмой грудной клетки у больных на фоне миксоматозной дегенерации хорд. Основным патогенетическим механизмом острой митральной недостаточности является легочная венозная гипертензия, возникающая из-за большого объема регургитации в недостаточно растяжимое левое предсердие. Клиническая симптоматика проявляется внезапным развитием отека легких.
У детей митральная недостаточность при ПМК протекает чаще всего бессимптомно и диагностируется при допплерэхокардиографическом исследовании. В последующем при прогрессировании регургитации появляются жалобы на одышку при физической нагрузке, снижение физической работоспособности, слабость, отставание в физическом развитии.
Факторами риска развития «чистой» (не воспалительной) митральной недостаточности при синдроме пролабирования по данным двухмерной эхокардиографии являются:
- Дилятация левого атриовентрикулярного отверстия.
- Пролапс преимущественно задней митральной створки.
- Утолщенность задней митральной створки.
ПМК является высоким фактором риска возникновения инфекционного эндокардита. Абсолютный риск возникновения заболевания выше, чем в популяции, в 4,4 раза.
Диагностика инфекционного эндокардита при ПМК представляет определенные трудности. Поскольку створки при пролапсе избыточно фестончатые, это не позволяет выявить начало формирования бактериальных вегетаций по данным эхокардиографии. Поэтому основное значение в диагностике эндокардита играют: 1) клиническая симптоматика инфекционного процесса (лихорадка, ознобы, сыпь, и другие симптомы), 2) появление шума митральной регургитации и факт обнаружения возбудителя при повторных высевах крови.
Частота внезапной смерти при синдроме ПМК зависит от многих факторов, основными из которых являются электрическая нестабильность миокарда при наличии синдрома удлиненного интервала QT, желудочковых аритмий, сопутствующая митральная недостаточность, нейрогуморальный дисбаланс.
Риск внезапной смерти при отсутствии митральной регургитации низкий и не превышает 2:10 000 в год, в то время как при сопутствующей митральной регургитации увеличивается в 50–100 раз.
В большинстве случаев внезапная смерть у больных с ПМК носит аритмогенный генез и обусловлена внезапным возникновением идиопатической желудочковой тахикардии (фибрилляции) или на фоне синдрома удлиненного интервала QT.
В редких случаях в основе внезапной сердечной смерти у больных с ПМК может лежать врожденная аномалия коронарных артерий (аномальное отхождение правой или левой коронарной артерии), приводящая к острой ишемии миокарда и его некрозу.
Таким образом, основными факторами риска внезапной смерти у детей с синдромом ПМК являются: желудочковые аритмии III–V градации по Lown; удлинение корригированного интервала QT более 440 мс; появление ишемических изменений на ЭКГ во время физической нагрузки; кардиогенные обморочные состояния в анамнезе.
ДСТС являются одними из неблагоприятных факторов, предрасполагающих к развитию аритмических осложнений в детском и подростковом возрасте, в том числе гемодинамически значимых. В структуре нарушений ритма у детей с ДСТС чаще выявляются наджелудочковая экстрасистолия в патологическом количестве и желудочковая экстрасистолия, взаимосвязанные со степенью кардиальной дисплазии (Гнусаев С. Ф., соавт., 2006).
Морфологическими проявлениями синдрома ДСТС у детей с сопутствующей патологией почек, по данным Домницкой Т. М., Гавриловой В. А. (2000), являются: шаровидная или треугольная форма сердца, закругление верхушки сердца, увеличение массы сердца в 1,4–2,5 раза, утолщение и укорочение хорд митрального клапана, отхождение хорд в виде веера, гипертрофия сосочковых мышц, воронкообразная форма митрального клапана, открытое овальное окно. Миксоматозная дегенерация створок атриовентрикулярных клапанов наблюдалась у большинства больных с синдромом ДСТС и заболеваниями органов мочевой системы (частота ее колебалась от 66,7% до 77%). Фиброэластоз эндокарда был выявлен у 10 детей анализируемой группы.
В популяции детей наиболее часто выявлялись смещение септальной створки трехстворчатого клапана в полость желудочка в пределах 10 мм, нарушенное распределение хорд передней створки митрального клапана, дилятация синусов Вальсальвы, увеличенная евстахиева заслонка более 1 см, дилятация ствола легочной артерии, ПМК, диагонально расположенные трабекулы в полости левого желудочка.
Тактика ведения детей с первичным ПМК различается в зависимости от степени выраженности пролабирования створок, характера вегетативных и сердечно-сосудистых изменений. Основными принципами лечения являются: 1) комплексность; 2) длительность; 3) учет направленности функционирования вегетативной нервной системы.
Обязательным является нормализация труда, отдыха, распорядка дня, соблюдение правильного режима с достаточным по продолжительности сном.
Вопрос о занятиях физкультурой и спортом решается индивидуально после оценки врачом показателей физической работоспособности и адаптивности к физической нагрузке. Большинство детей при отсутствии митральной регургитации, выраженных нарушений процесса реполяризации и желудочковых аритмий удовлетворительно переносят физическую нагрузку. При наличии врачебного контроля им можно вести активный образ жизни без каких-либо ограничений физической активности. Детям можно рекомендовать плавание, лыжи, коньки, катание на велосипеде. Не рекомендуются спортивные занятия, связанные с толчкообразным характером движений (прыжки, борьба каратэ и др.). Обнаружение у ребенка митральной регургитации, желудочковых аритмий, изменений обменных процессов в миокарде, удлинения интервала QT диктует необходимость ограничения физической активности и занятий спортом. Этим детям разрешается занятие лечебной физкультурой под контролем врача.
Лечение строится по принципу общеукрепляющей и вегетотропной терапии. Весь комплекс терапевтических мероприятий должен строиться с учетом индивидуальных особенностей личности больного и функционального состояния вегетативной нервной системы.
Важной частью комплексного лечения детей с ДСТС является немедикаментозная терапия: психотерапия, аутотренинг, физиотерапия (электрофорез с магнием, бромом в области верхнешейного отдела позвоночника), водные процедуры, иглорефлексотерапия, массаж позвоночника. Внимание врача должно быть направлено на санацию хронических очагов инфекции, по показаниям проводится тонзиллэктомия.
Медикаментозная терапия должна быть направлена на: 1) лечение вегетативно-сосудистой дистонии; 2) предупреждение возникновения нейродистрофии миокарда; 3) психотерапию; 4) антибактериальную профилактику инфекционного эндокардита.
При умеренных проявлениях симпатикотонии назначается фитотерапия седативными травами, настойка валерианы, пустырника, сбор трав (шалфей, багульник, зверобой, пустырник, валериана, боярышник), обладающий одновременно легким дегидратационным эффектом. При наличии изменений процесса реполяризации на ЭКГ, нарушениях ритма проводятся курсы лечения препаратами, улучшающими обменные процессы в миокарде (панангин, карнитин, Кудесан, витамины). Карнитин назначают в дозе 50 мг/кг в сутки на 2–3 мес. Карнитин выполняет центральную роль в липидном и энергетическом обмене.
Являясь кофактором бета-окисления жирных кислот, он переносит ацильные соединения (жирные кислоты) через митохондриальные мембраны, предупреждает развитие нейродистрофии миокарда, улучшает его энергетический обмен. В наших исследованиях 35 детям с экстрасистолией (более 15 в одну минуту) в состав комплексной терапии был включен карнитин. По окончании лечения у 25 детей экстрасистолия значительно уменьшилась, у 10 детей — не определялась.
Отмечен благоприятный эффект от применения препарата Коэнзим Q10®, который значительно улучшает биоэнергетические процессы в миокарде и особенно эффективен при вторичной митохондриальной недостаточности.
Ранняя диагностика ДСТ у детей позволяет осуществлять соответствующую реабилитационную терапию и предотвращать прогрессирование заболевания. Одним из наиболее ярких терапевтических результатов является эффективное лечение детей с ДСТ (главным образом с ПМК) при помощи магнийсодержащего препарата магния оротата — Магнерот®. Выбор препарата был обусловлен известными свойствами иона магния, отмечающимися у антиаритмических препаратов I и IV класса (мембраностабилизирующие и антагонисты кальция), а также отсутствием побочных эффектов, которые могут появляться при применении традиционной антиаритмической терапии. Учитывалось также и то, что действующим веществом препарата является магния оротат, который, индуцируя синтез протеинов, участвуя в обмене фосфолипидов, являющихся составной частью клеточных мембран, необходим для фиксации внутриклеточного магния (Громова О. А., 2007).
Препарат Магнерот® применялся в виде монотерапии в дозе 40 мг/кг в сутки в течение первых 7 дней приема, затем по 20 мг/кг в сутки в течение 6 месяцев. Результатом лечения явилось уменьшение на 20–25% глубины пролабирования створок митрального клапана и уменьшение степени регургитации на 15–17%. Терапия препаратом Магнерот® не влияла на размеры левых отделов сердца и сократимость миокарда, показатели которых до лечения находились в пределах нормы.
В исследованиях, проведенных Е. Н. Басаргиной (2008), выявлен антиаритмический эффект препарата Магнерот®. При проведении суточного мониторирования ЭКГ у детей 2-й и 3-й групп было отмечено уменьшение количества желудочковых комплексов на 50% и более у 18 (27,7%) больных. Причем у 6 детей отмечено исчезновение желудочковой аритмии или уменьшение количества желудочковых комплексов до 30–312 за сутки. У 14 (21,5%) детей количество желудочковых комплексов уменьшилось не менее чем на 30%. У двух больных отмечено увеличение количества желудочковых экстрасистол до 30% от исходного уровня. Таким образом, антиаритмическая эффективность препарата Магнерот® составила 27,7%. Подобные результаты ранее получены и в других исследованиях (Домницкая Т. М. и соавт., 2005).
В то же время редкие суправентрикулярные и желудочковые экстрасистолы, если не сочетаются с синдромом удлиненного интервала QT, как правило, не требуют назначения каких-либо антиаритмических препаратов.
Таким образом, дети с синдромом ДСТС нуждаются в своевременной диагностике с использованием допплерэхокардиографии, электрокардиографии, в ряде случаев суточного мониторирования ЭКГ, назначении индивидуальной терапии и наблюдении детским кардиологом.
Терапия препаратом Магнерот® у детей с синдромом ДСТС приводит к уменьшению признаков пролапса клапанов, частоты выявления митральной регургитации, уменьшению выраженности клинических проявлений вегетативной дисфунуции, частоты желудочковых аритмий, сопровождается повышением уровня внутриэритроцитарного магния.
Литература
- Земцовский Э. В. Диспластические синдромы и фенотипы. Диспластическое сердце. Спб: «Ольга». 2007. 80 с.
- Гаврилова В. А. Синдром дисплазии соединительной ткани сердца у детей с заболеваниями органов мочевой системы. Автореф. дисс. д.м.н. М., 2002.
- Morales A. B., Romanelli B., Boucek R. J. et al. Myxoid heart disease: an assessment of extravalvular cardiac pathology in severe mitrae valve prolapse // Hum.Pathol. 1992, v. 23, № 2, p. 129–137.
- Верещагина Г. Н. Системная дисплазия соединительной ткани. Клинические синдромы, диагностика, подходы к лечению. Методическое пособие для врачей. Новосибирск, 2008, 37 с.
- Урмонас В. К., Кондрашин Н. И. Воронкообразная грудная клетка. Вильнюс: Мокслас, 1983, 115 с.
- Гнусаев С. Ф. Значение малых аномалий сердца у здоровых детей и при сердечно-сосудистой патологии. Автореф. дисс. д.м.н., М., 1996.
- Белозеров Ю. М., Гнусаев С. Ф. Пролапс митрального клапана у детей. М.: Мартис, 1995. 120 с.
- Сторожаков Г. И., Верещагина Г. С., Малышева Н. В. Оценка индивидуального прогноза при пролапсе митрального клапана // Кардиология, 2004, 4, с. 14–18.
- Нечаева Г. И., Викторова И. А. Дисплазия соединительной ткани: терминология, диагностика, тактика ведения пациентов. Омск: Изд-во «Типография Бланком», 2007. 188 с.
- Гнусаев С. Ф., Белозеров Ю. М., Виноградов А. Ф. Клиническое значение малых аномалий сердца у детей // Российский вестник перинатологии и педиатрии. 2006, № 4. С. 20–24.
- Домницкая Т. М., Гаврилова В. А. Синдром дисплазии соединительной ткани сердца у детей с заболеваниями мочевой системы / Материалы Второго Съезда педиатров-нефрологов России. М., 2000. С. 159.
- Громова О. А, Гоголева И. В. Применение магния в зеркале доказательной медицины и фундаментальных исследований в терапии // Фарматека. 2007, т. 146, № 12, с. 3–6.
- Басаргина Е. Н. Синдром дисплазии соединительной ткани сердца у детей // Вопросы современной педиатрии. 2008, т. 7, № 1, 129–133.
- Домницкая Т. М., Дьяченко А. В., Куприянова О. О., Домницкий М. В. Клиническая оценка использования оротата магния улиц молодого возраста с синдромом дисплазии соединительной ткани сердца // Кардиология. 2005; 45 (3): 76–81.
С. Ф. Гнусаев, доктор медицинских наук, профессор
ГОУ ВПО Тверская ГМА Росздрава, Тверь
Контактная информация об авторе для переписки
2.Причины
Генетический, наследственный характер заболевания распространяется лишь на 30-50% случаев, причем уже идентифицированы ответственные за его развитие гены. В региональном варианте АКМП ПЖ, известном как болезнь острова Наксос (Греция), фиброзно-жировая дегенерация и прогрессирующая сердечная недостаточность сопровождаются врожденными аномалиями кожи и волос (кератодермия, «шерстяные волосы»).
Этиопатогенез всех прочих, ненаследственных случаев остается, по сути, неизвестным. Рассматриваются и обсуждаются гипотезы, согласно которым дегенеративный процесс в правом желудочке миокарда запускается вследствие бактериального или вирусного инфекционного воспаления; идиопатическими (сугубо индивидуальными) особенностями внутриутробного развития; аномально ускоренным апоптозом (преждевременной гибелью кардиомиоцитов после значительно меньшего, чем в норме, числа клеточных делений); спонтанным перерождением кардиомиоцитов в клетки других типов (трансдифференциация). Однако даже если какое-либо из этих предположений подтвердится статистически, оно мало чего будет стоить без понимания внутренних причин: почему у одних людей аритмогенная дисплазия запускается и развивается, а у других нет.
Посетите нашу страницу Кардиология
Симптомы появления аритмогенной дисплазии правого желудочка
Иногда болезнь долго протекает бессимптомно, и первым ее проявлением может стать внезапная смерть.
В других случаях пациента беспокоят эпизоды учащенного сердцебиения и обморочные состояния, вызванные пароксизмальной желудочковой тахикардией. При менее опасной желудочковой экстрасистолии иногда возникает ощущение перебоев в сердце.
Дети и подростки могут жаловаться на головокружения, неожиданные приступы сердцебиения или резкой слабости, быструю утомляемость.
При прогрессировании правожелудочковой недостаточности появляются отеки на ногах, тяжесть в правом подреберье, увеличение живота, слабость. При нарушении функции левого желудочка присоединяется одышка при физической нагрузке, приступы удушья в положении лежа, кашель.
3.Симптомы и диагностика
Клиническая картина АКМП ПЖ требует дифференциации с множеством иных заболеваний и состояний, при которых постепенно прогрессирующая сердечная недостаточность сочетается с эпизодами аритмии. Как правило, пациенты начинают обращать внимание на усиленное сердцебиение, перебои сердечного ритма, одышку, затем появляются боли в сердце, головокружения, внезапные синкопальные состояния с потерей сознания (прогностически неблагоприятный признак, который считается одним из возможных предвестников внезапной сердечной смерти). Нарастает утомляемость, пациент все хуже переносит физические нагрузки.
Однако во многих случаях прогрессирование фиброзно-жировой дегенерации в течение длительного времени не сопровождается сколько-нибудь значимыми субъективными ощущениями или клиническими симптомами.
Учитывая диагностическую сложность АКМП ПЖ, международные кардиологические ассоциации предпринимали попытки не только унифицировать терминологию, но и разработать надежные протоколы распознавания и дифференциальной диагностики. Так, необходимым для установления диагноза считается присутствие не менее 3% соединительной и не менее 40% жировой ткани в гистологической структуре правого желудочка; применяются также электрокардиографические, аускультативные и другие диагностические критерии. В силу очевидных причин требуется изучение семейного анамнеза.
По мере необходимости назначают ЭЭГ, эхокардиографию, нагрузочные тесты, холтеровское мониторирование, рентгенографию, МРТ, а если все эти методы не позволяют однозначно определиться с диагнозом – приходится прибегать к инвазивной эндомиокардиальной биопсии, отбирая тканевые образцы для гистологического анализа.
О нашей клинике м. Чистые пруды Страница Мединтерком!
Что происходит в органе при болезни Фонтана
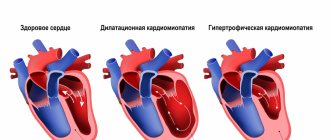
Структурные нарушения при этом заболевании происходят в результате липидного пропитывания и разрастания соединительной ткани в мышце правого желудочка. Это ведет к его прогрессирующему расширению и снижению сократимости. Левый желудочек поражается значительно реже, межжелудочковая перегородка практически не вовлекается. Вовлечение в процесс и левого желудочка ассоциировано с плохим прогнозом заболевания.
Механизмы разрушения клеток миокарда:
- апоптоз (запрограммированная смерть клетки);
- воспаление, усиливающийся фиброз и потеря способности к сокращению;
- жировое замещение клеток сердца.
Аритмогенная дисплазия правого желудочка, аутопсия сердца молодого человека, который внезапно умер во время игры в баскетбол.
В результате нарушается электрическая стабильность миокарда, в нем появляются очаги патологического возбуждения. Они становятся источником желудочковых аритмий.
Желудочковая тахикардия, возникшая внезапно, может стать причиной сердечной смерти у молодого и внешне здорового человека.
При дальнейшем замещении миокарда жировой и соединительной тканью страдает функция правого желудочка, развивается его недостаточность. На конечных стадиях болезни имеется уже недостаточность обоих желудочков, и заболевание приобретает черты другой кардиомиопатии — дилатационной.
4.Лечение
Этиопатогенетической терапии в настоящее время не существует, и она не появится до окончательного прояснения всех вопросов, связанных с причинами и патогенезом аритмогенной правожелудочковой дисплазии. Риск внезапной фибрилляции и остановки сердца снижают бета-адреноблокаторами и другими антиаритмическими средствами; симптоматически назначают сердечные гликозиды, мочегонные, ингибиторы АТФ и т.д. Эффективность таких тактических схем требует дополнительных исследований. Обязательно вносятся коррективы в образ жизни, категорически исключаются любые факторы риска и факторы-провокаторы. В качестве лечения некоторыми кардиоцентрами практикуется радиочастотная катетерная абляция, однако этот опыт также пока изучен недостаточно.
Высказываются мнения о том, что методом выбора следует признать радикальное вмешательство – пересадку сердца или, по меньшей мере, вживление кардиовертера-дефибриллятора, – однако другим специалистам риск возможных осложнений при транс- или имплантации представляется оправданным лишь в тех случаях, когда процесс прогрессирует быстро и проявляется заведомо угрожающей симптоматикой.
Диагностика аритмогенной дисплазии правого желудочка
Диагноз ставится при наличии определенных критериев. Для их обнаружения необходимы такие исследования:
- ЭхоКГ;
- МРТ сердца;
- биопсия эндокарда и миокарда;
- радиоизотопная ангиография;
- ЭКГ;
На ЭКГ больного АДПЖ: отрицательные зубцы Т в отведениях V1–V5, стрелками указаны эпсилон-волны.
- суточное мониторирование ЭКГ;
- электрофизиологическое исследование сердца;
- трехмерное ЭКГ-картирование;
- молекулярно-генетическое тестирование.
ЭхоКГ при аритмогенной дисплазии правого желудочка смотрите в этом видео:
«Золотой стандарт» диагностики – рентгенконтрастная вентрикулография. Во время этого исследования в полости сердца через вену вводится вещество, непроницаемое для рентгеновских лучей. Характерными признаками заболевания являются:
- расширение (дилатация) правого желудочка;
- нарушение сократимости отдельных участков миокарда;
- выпячивание в зоне пораженных кардиомиоцитов;
- усиление трабекулярности – увеличение числа мышечных волокон в полости возле стенок желудочка.
Многие из этих методов исследования проводятся лишь в специализированных кардиологических центрах. Поэтому важно вовремя заподозрить болезнь по клиническим данным, и при появлении необъяснимых головокружений, обмороков, приступов сердцебиения обратиться к врачу.
КРИТЕРИИ ДИАГНОСТИКИ АДПЖ
Лечение болезни Фонтана
Учитывая две главные опасности патологии – аритмию и сердечную недостаточность, врачи применяют 2 направления терапии:
- назначаются антиаритмические препараты для предотвращения желудочковой тахикардии: соталол, амиодарон или флекаинид;
- при сердечной недостаточности применяются карведилол, ингибиторы АПФ, при отеках – диуретики, антагонисты альдостерона.
При выраженном замедлении пульса, в том числе вызванном приемом антиаритмических препаратов, возможна установка кардиостимулятора. Это устройство поддерживает сокращения сердца при замедлении пульса.
При неэффективности медикаментозной терапии может быть установлен кардиовертер-дефибриллятор или выполнена радиочастотная абляция источника желудочковой тахикардии. Последний вариант используется достаточно редко, так как его эффективность именно при этом заболевании невысока.
Кардиовертер-дефибриллятор устанавливают пациентам с расширением и снижением сократимости правого желудочка, вовлечением левого желудочка, наличием желудочковых тахикардий (по данным ЭФИ или ХМ ЭКГ). Это устройство обычно имплантируют молодым людям, и оно практически в 100% случаев помогает предотвратить у них внезапную смерть, вызванную приступом аритмии.
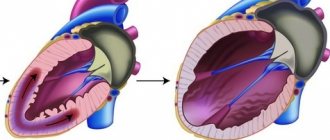
У больных, имеющих тяжелые нарушения ритма в сочетании с выраженной сердечной недостаточностью, проводится операция вентрикулотомия. Во время вмешательства происходит анатомическое прерывание путей, по которым идет аномальное возбуждение сердца при аритмии.
Наиболее эффективна трансплантация сердца, но эта операция выполняется редко.
Рекомендую прочитать об открытом артериальном протоке. Вы узнаете об этиологии и нарушении гемодинамики при ОАП, симптомах заболевания, диагностических мероприятиях и лечении. А здесь подробнее о синдроме Вольфа-Паркинсона-Уайта.
Этиология
Аритмогенная кардиомиопатия правого желудочка относится к категории редких, потому что диагностируется у 1–6 людей на 10 тысяч населения. Большинство пациентов составляют представители мужского пола — у мужчин болезнь встречается в 4 раза чаще, чем у женщин.
Предрасполагающие факторы, почему развивается аритмогенная кардиомиопатия, остаются неизвестными для специалистов из области кардиологии. Однако клиницистами выдвинуто несколько теорий касательно происхождения. Основная — генетическая предрасположенность.
Считается, что заболевание передается по аутосомно-доминантному типу, а мутантный ген располагается в 12, 14, 17 и 18 хромосомах, которые блокируют специфические белки, выделяемые миокардом. Распространенность такого источника заболевания составляет от 30 до 50 %.
Среди теорий происхождения заболевания стоит выделить такие:
- Метаболическая. Процесс дисплазии может быть связан с метаболическими нарушениями, протекающими в миокарде правого желудочка. Основой подобного предположения выступает болезнь под названием мышечная дистрофия скелетных мышц с постоянным прогрессированием. Однако неизвестно, почему дисплазия не оказывает негативного влияния на костную систему.
- Воспалительная. Основана на том, что примерно у 80 % пациентов с подобным диагнозом во время гистологического изучения миокарда была обнаружена воспалительная инфильтрация. Авторы теории уверены, что заболевание — следствие протекания миокардита.
- Верифицированный апоптоз (запрограммированная смерть клетки). Несмотря на существование такого предположения, остается неизвестным, почему степень апоптоза выше в правом желудочке и только при отсутствии терапии распространяется на сердечную перегородку с последующим повреждением левого желудочка.
В единичных случаях заболевание наследуется по аутосомно-рецессивному типу — эта разновидность носит название болезнь Наксоса. В настоящее время лишь 25 человек в мире живет с подобной патологией.