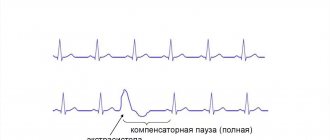
In Latin there is a word compensatum, which means “to balance.” Compensatory pause is a term that characterizes the diastolic pause that occurs after a heart rhythm disturbance. In terms of time, such a pause is extended. Its duration is equal to two pauses normal for heart rhythm.
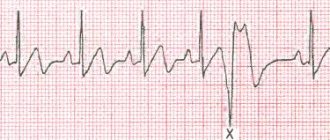
A compensatory pause occurs after the ventricular extrasystole and lasts until the next independent contraction.
Reasons for the occurrence of a compensatory pause
After a ventricular extrasystole, a refractory period is observed, characterized by the fact that the ventricle does not respond to the next impulse emanating from the sinus. This leads to the fact that the ventricle contracts not after the first, but after the second sinus impulse. There are cases when the heartbeat is very rare, the end of the refractory period is observed after the extrasystole and before the next sinus impulse. Such changes in heart rhythm can lead to a lack of compensatory pause.
Heart rhythm can be nomotopic or heterotopic. Their simultaneous presence in a person is called parasystole, which can often cause compensatory pauses.
Another reason for their appearance may be extrasystolic allorhythmia, which is a serious pathology associated with impaired circulatory function and heart rhythm.
Long and short QT syndrome in clinical practice
Among the causes of prolongation and shortening of the QT interval, congenital and acquired factors are distinguished. The main cause of the disease is hereditary channelopathy, caused by mutations in a number of genes encoding proteins of transmembrane potassium and sodium ion channels. Long QT syndrome (LQS) has a fairly long history of study, dating back more than 100 years. Perhaps the first description of the hereditary QT SUDI is the work of T. Messner et al., published in 1856 [1]. A powerful impetus for the study of cardiac electrophysiology was the introduction into medical practice of the ECG recording technique developed by the Dutch physiologist V. Eindhoven in 1903. But only in 1957, A. Jervell and F. Lange-Nielsen diagnosed clinical electrocardiographic “long QT syndrome” in four members of the same family who suffered from congenital neural deafness, frequent attacks of loss of consciousness and had persistent prolongation of the QT interval on the ECG, which marked the beginning of the modern stage of studying QT JUDG. Soon P. Romano (1963) and D. Ward (1964) presented observations of a similar syndrome, but without congenital deafness. At the same time, relatives of patients also showed prolongation of the QT interval, but no attacks of loss of consciousness were observed [1]. The arrhythmogenic potential of a short QT interval was first noted by I. Gussak et al. in 2000, when describing a clinical case of sudden cardiac death of a young woman and a family in which several cases of early onset atrial fibrillation (AF) were observed among its members. None of the subjects had structural changes in the heart, but there was a clear decrease in the duration of the QT interval on the ECG (QTC ranged from 248 to 300 ms) [2].
Electrophysiology of the cardiac cell and the relationship with the duration of the QT interval The QT interval of the ECG reflects the total duration of depolarization and repolarization of ventricular cardiomyocytes. At the level of an individual cell, the QT interval corresponds to the duration of the transmembrane action potential (TMAP), caused by the transmembrane flow of ions through sodium, calcium and potassium channels. There are five successive phases of TMPD: Phase 0 (depolarization) is characterized by a massive flow of sodium ions into the cell (INa). Phase 1 (initial rapid repolarization) is characterized by cessation of sodium ion flow and a transient fast flow of potassium ions out of the cell (It0). Phase 2 (plateau) is characterized by a slow flow of calcium ions into the cell through L-type calcium channels (ICa-L) and a continued flow of potassium ions outward (IK). Phase 3 (final rapid repolarization) is characterized by the flow of potassium ions out of the cell (IKr, IKs) with the formation of the resting transmembrane potential (RTMP). Phase 4 (depolarization) is characterized by the maintenance of TMPP due to the active entry of potassium ions into the cell (IK1). At the microstructural level, transmembrane ion channels are complex structural formations consisting of specific protein complexes. Dysfunction of these protein channels may cause transmembrane ion fluxes to accelerate or decelerate during various phases of TMPD, which may result in prolongation or shortening of TMPD duration and the QT interval. The main cause of dysfunction of transmembrane ion channels is mutation of the genes encoding their proteins. Mutations can affect all types of channels, as well as their combinations, which determines the existence of a large number of clinical forms of long and short QT interval syndrome. Currently, the structure and genetics of transmembrane ion channels have been fully studied, which makes drug correction of their disorders accessible. Detailed literature on this issue is presented in the review by S. Nachimuthu et al. [3].
Methodology for measuring and assessing the QT interval The QT interval is measured on the ECG from the beginning of the Q wave (if it is absent, from the beginning of the R wave) to the end of the T wave. Despite its apparent simplicity, measuring and assessing the QT interval is a rather difficult task and is one of the the most difficult moments in ECG analysis. The greatest difficulties are: 1) determining the beginning of the QRS complex and the end of the T wave; 2) selection of leads in which it is preferable to measure the QT interval; 3) the need to adjust the duration of the QT interval for heart rate, gender and duration of the QRS complex [4].
According to a number of studies, in healthy people in different leads the duration of the QT interval can vary within 50–65 ms. According to the 2009 American Heart Association ECG Standardization and Interpretation Guidelines [4], when measuring QT interval in individual leads, the lead with the longest QT interval (usually lead V2 or V3) should be selected for analysis. In most cases, the end of the T wave is determined at the moment the final part of the T wave returns to the isoline. In the case of a “two-humped” T wave with peaks of equal amplitude, it is recommended to determine the end of the T wave by the end of the second peak [5]. If the T and U waves overlap, it is recommended to measure the QT interval in leads without a U wave (often leads aVR or aVL) or determine the end of the T wave at the intersection of the isoline with a line drawn tangentially along the descending part of the T wave (necessary take into account that the latter method may underestimate the QT interval) [4] (Fig. 1). When measuring manually, it is recommended to determine the duration of the QT interval as the average value of several measurements (at least 3–5 cardiac cycles) [5]. In recent years, many modern electrocardiographs have become capable of automated ECG analysis, including determination of the duration of the QT interval. Superposition and averaging of several leads used in automatic analysis allow more accurate determination of the beginning and end of the QT interval, as a result of which the automatically measured QT interval is often longer than the QT interval with the manual measurement method. Therefore, if QT interval prolongation is detected during automated analysis, it is recommended to recheck the results manually [4]. It is known that the duration of the QT interval has a clear relationship with heart rate (RR interval): when the heart rate decreases, the QT interval increases, and when the heart rate increases, it decreases. This feature indicates the need to correct the duration of the QT interval depending on heart rate. For this purpose, a number of formulas have been proposed using exponential, linear or logarithmic methods [5]. It should be noted that in the heart rate range from 60 to 90 beats/min. Most formulas provide comparable correction results and are interchangeable.
One of the first formulas for correcting the QT interval depending on heart rate was proposed by HC Bazett in 1920, and to this day it remains the main formula for determining the corrected QT interval (QTc) in both research and clinical practice. Most electrocardiographs use the Bazett formula for automated analysis. The Bazett formula uses the exponential method to determine QTc (QTc=QT/RR1/2). The disadvantages of the Bazett formula include the possibility of erroneous correction if the heart rate is too high or low. Formulas using the linear correction method (Framingham, Hodges, Rautaharju) can reduce the errors of the exponential method and can be used for both high and low heart rates. The most famous of them is the Framingham formula (QTc=QT + 0.154 x (1 – RR)), and the most accurate, but more complex, is the Rautaharju formula. Details of various methods for correcting the QT interval for heart rate can be found in the review by I. Goldenberg et al. [5].
It should be noted that manual determination of QTc for each individual patient is a rather labor-intensive and time-consuming process. Therefore, in clinical practice, a QT-HR nomogram can be used to quickly identify patients at risk for torsades de pointes (TdP) [3]. Since the QT interval can increase with intraventricular conduction disorders, to assess the duration of repolarization in patients with bundle branch blocks, it is recommended to use either the duration of the JT interval (from the beginning of the ST segment to the end of the T wave) or correction formulas that take into account both heart rate and the duration of the QRS complex [ 4]. Unfortunately, these methods of analysis still do not have generally accepted standards and are used very limitedly in clinical practice.
QT interval: long, normal, shortened In 2009, S. Viskin [6], using data from population and genetic studies, developed a “QT scale” that ranks the entire continuous spectrum of QT intervals from very short to very long, separately for men and women . In accordance with this scale, the normal duration of the QT interval is considered to be QTc values of 360–389 ms for men and 370–399 ms for women; with a QTc equal to 390–449 ms for men and 400–459 ms for women, the QT interval was regarded as possibly prolonged; with QTc equal to 450–469 ms for men and 460–479 ms for women, as prolonged; with a QTc equal to or greater than 470 ms for men and 480 ms for women, as markedly prolonged; with a QTc equal to 359–329 ms for men and 369–339 ms for women, as shortened, with a QTc equal to or less than 330 ms for men and 340 ms for women, as markedly shortened. One of the first and most well-known criteria for diagnosing QT SUDS are the criteria of PJ Schwartz et al. 1985, which were subsequently supplemented and updated several times (Table 1). In accordance with these criteria, persons scoring 1 point have a low probability of a QT JUDG, from 2 to 3 points - an intermediate probability, 4 points or more - a high probability of a QT JUDG [7]. In 2011, MH Gollob et al. [2] proposed diagnostic criteria for short QT interval syndrome (SQTS), based on the same principles as the criteria for QT QTS (Table 2). In accordance with these criteria, if the total number of points is 4 or more, a high probability of QT BITCH is determined; if there are 2 points or less, a low probability is determined; if the total score is 3 points, then the probability of QT BITCH is assessed as intermediate.
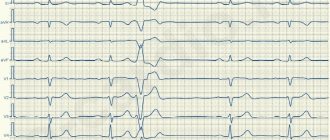
Long QT interval syndrome An increase in the duration of repolarization often leads to the appearance of intense oscillations on the membrane of ventricular cardiomyocytes, called early afterdepolarization potentials, which, in combination with the pronounced heterogeneity of the duration of action potentials, causes the occurrence of foci of repeated excitation and ventricular tachycardia in the ventricular myocardium [3]. The most characteristic clinical manifestation of QT VSD is polymorphic ventricular tachycardia torsades de pointes (TdP) (bidirectional, “pirouette” tachycardia). TdP is characterized by a pronounced prolongation of the QT interval in the last sinus contraction preceding tachycardia, a progressive change in the polarity of the QRS complexes, visually simulating their rotation around the isoline, a constant change in the amplitude of the QRS complexes, a high heart rate from 150 to 300 pulses per minute and pronounced irregularity of the RR intervals (Fig. 2 ). TdP is characterized by the onset of an attack after a pause caused by bradycardia or extrasystole. Typical for TdP is the so-called SLS (short-long-short) sequence, characterized by an initial supraventricular extrasystole leading to a shortening of the RR interval (short cycle), followed by a long post-extrasystolic pause before the next sinus complex (long cycle) and repeated ventricular extrasystole ( short cycle), which is the beginning of TdP paroxysm. In patients with QT VSD, the occurrence of TdP is often provoked by intense adrenergic stimulation [8]. Attacks of TdP in patients with QT SUDS are usually short-lived, resolve spontaneously, and therefore can go undetected for a long time. However, these attacks tend to cluster in repeated sequences with short interictal intervals, causing palpitations, dizziness, syncope, presyncope, and sudden death due to ventricular fibrillation (VF).
In recent decades, significant progress has been made in determining the genetic prerequisites for the occurrence of QT. Mutations in ten genes have been identified that cause prolongation of the QT interval. However, the vast majority of clinically significant cases of LQT QT are associated with mutations in three genes, manifesting in three genetic subtypes (LQT1, LQT2 and LQT3), which have characteristic clinical features and are characterized by specific ECG morphology [9]. LQT1 is characterized by wide T waves on the resting ECG, the absence of a pause before the onset of tachyarrhythmia, the absence of shortening of the QT interval during exercise, and the high effectiveness of β-blockers (BAB). The development of tachyarrhythmia in LQT1 is provoked by physical and mental stress, swimming, and diving. LQT2 is characterized by low-amplitude, jagged T waves on the resting ECG, the presence of a pause before the onset of tachyarrhythmia, normal shortening of the QT interval during exercise, and lower effectiveness of beta blockers compared to LQT1. The development of tachyarrhythmia in LQT2 is provoked by physical and mental stress, sudden loud sounds. LQT3 is characterized by a long isoelectric ST segment, narrow and high T waves on the resting ECG, and excessive shortening of the QT interval during exercise. The effectiveness of beta blockers has not been determined. Tachyarrhythmia most often occurs at rest, during sleep.
Most cases of QT JSD are represented by autosomal dominant forms with varying penetrance. Mutations are more often detected in individuals whose parents themselves are carriers of mutant genes. In women, the risk of syncope and sudden death decreases during pregnancy, but increases again in the postpartum period. Fainting and sudden death are observed mainly in children and adolescents and are not typical for people over 40 years of age [9]. The frequency of mutations in genes responsible for prolongation of the QT interval is approximately 1 in 2 thousand people, but the frequency of manifest forms is significantly lower, since most carriers of defective genes have no symptoms throughout their lives.
Diagnosis of hereditary QT VSD is based on identifying characteristic ECG changes, analysis of clinical data and family history in accordance with the criteria of PJ Schwartz et al. [7], as well as excluding acquired causes of QT interval prolongation. The final stage of diagnosis is genetic testing, which makes it possible to identify the affected gene in 70–90% of individuals with signs of hereditary QT. Despite its high diagnostic value, genetic testing is not a panacea and can give both false-positive and false-negative results. Genetic testing is indicated mainly in two cases: 1) when the diagnosis is probable and clinical data indicate damage to a specific gene; 2) in families in which there is a proband with a previously established genetic defect. In both cases, genetic testing is necessary to clarify the diagnosis, determine the prognosis and choose long-term treatment tactics [9]. In recent years, a large number of non-hereditary factors causing prolongation of the QT interval and TdP have been identified, primarily drugs, including class Ia antiarrhythmics (quinidine, procainamide, disopyramide) and class III (dofetilide, ibutilide, sotalol), antipsychotics (haloperidol, droperidol, thioridazine, chlorpromazine), antidepressants (amitriptyline, desipramine, imipramine, maprotiline, doxepin, fluoxetine), quinolone antibiotics (levofloxacin, moxifloxacin) and macrolides (erythromycin, clarithromycin), antimalarials (quinidine), antiprotozoal drugs ( pentamidine), antifungals (azole group) and methadone [3]. At the same time, the prognostic value of acquired prolongation of the QT interval has not been sufficiently studied. It is noted that the relationship between the mechanism of action of the drug agent and the clinical manifestations of LDS QT is not strict. In some cases, even significant prolongation of the QT interval is rarely accompanied by the development of TdP (for example, with the use of amiodarone), and in others, a slight prolongation of the QT interval can cause TdP [3].
It is suggested that the likelihood of developing TdP increases when multiple risk factors are combined. The main risk factors for acquired QT TSD include electrolyte disturbances (hypokalemia, hypomagnesemia, hypocalcemia), the use of drugs that prolong the QT interval, diuretics and cardiac glycosides, the presence of concomitant diseases (liver and renal failure, bradycardia, heart failure, left ventricular hypertrophy, infarction myocardium, subarachnoid hemorrhage and other forms of damage to the central nervous system), a diet with liquid proteins and other forms of fasting [3, 11]. It is known that 5 to 20% of patients with drug-related TdP have mutations in the genes that cause QT TDS. These patients typically have a normal or borderline QTc, but are prone to QT prolongation and TdP when exposed to certain medications, stress, or other risk factors [3].
Short QT syndrome QT is characterized by a hereditary shortening of the QT interval, accompanied by a high incidence of AF (24%) in the form of permanent or paroxysmal forms, frequent fainting, the development of polymorphic ventricular tachycardia, VF, cardiac arrest and sudden death. Depression of the PR segment, high peak-shaped T waves without horizontal flattening of the ST segment, impaired shortening of the ST segment with an increase in heart rate, and paradoxical shortening of the QT interval with bradycardia may also be observed. AF and VF in patients with QT stenosis are easily provoked by programmed cardiac pacing [11]. The electrophysiological basis for shortening the QT interval is a decrease in the duration of TMPD due to a decrease in depolarization currents (INa, ICa), an increase in repolarization currents (Ito, IK1, IK-ATP, IACh, IKr, IKs) or a combination thereof. Experimental studies show that the shortening of TMPP in patients with QT is characterized by pronounced heterogeneity, accompanied by transmural dispersion of repolarization, which is a substrate for the development of arrhythmias according to the “reentry” mechanism [12].
Currently, five genetic subtypes of short QT syndrome (SQT1–5) with autosomal dominant transmission have been described, associated with mutations in five different genes encoding potassium and calcium transmembrane ion channels (IKr, IKs, IK1, ICa). For SQT1 and SQT3–5, familial cases have been proven; SQT2 is described in a single sporadic case [12]. In SQT1, the provoking factor for heart rhythm disturbances is usually physical activity and loud noises; in SQT3, sudden awakening at night [11]. In addition to hereditary forms, shortening of the QT interval in clinical practice is most often found in hypercalcemia caused by hyperparathyroidism, kidney disease, osteolytic forms of cancer, taking thiazide diuretics, lithium and vitamin D. Among other clinical situations associated with secondary shortening of the QT interval, Brugada syndrome should be noted , chronic fatigue syndrome, hyperthermia, early ventricular repolarization syndrome, acidosis, the influence of digitalis, atropine and catecholamines [12, 13]. Secondary shortening of the QT interval increases the risk of arrhythmogenic events [13].
Treatment The lack of multicenter randomized controlled trials of treatment for long and short QT syndromes reflects both the relative rarity of these diseases and the large number of genetic types with significant differences in clinical features and severity. Patients at very low risk of sudden death (eg, elderly mutation carriers with normal QT interval duration) usually do not require treatment but should avoid drugs that prolong the QT interval [9].
The mainstay of treatment for patients with QT-LDS is beta blockers and implantable cardioverter defibrillators (ICDs) [9]. The main therapeutic effect of beta blockers is to prevent an increase in heart rate during physical activity and stress. Treatment of beta-blockers in patients with QT VSD is carried out according to generally accepted regimens, taking into account all possible contraindications. There is evidence that beta blocker therapy is more effective in patients with LQT1 than in patients with LQT2 and LQT3 [9]. A therapeutic effect comparable to BAB in patients with QT VSD is achieved with left cervical sympathectomy (LSS) (ganglionectomy of the stellate ganglion). Considering that LSS is an invasive operation, it is indicated for patients who have contraindications to beta blockers [14].
ICDs are widely used to prevent life-threatening arrhythmias and sudden death in patients with QT VSD. The main population for the treatment of CDI are: 1) individuals whose symptoms develop at an early age before the onset of puberty; 2) patients with a significantly prolonged QT interval (QTc>500 ms); 3) patients with repeated arrhythmogenic syncope that occurs during treatment with beta blockers. The issue of more aggressive tactics of ICD implantation in all carriers of mutant genes identified during family genetic screening remains controversial [9]. The joint recommendations of the North American and European Societies of Cardiology for the treatment and prevention of sudden death in QT SCD are presented in Table 3. ICD implantation is strongly recommended in all patients with QT SCD for secondary prevention of sudden cardiac death, unless there are absolute contraindications or patient refusal. At the same time, the use of ICDs for the primary prevention of sudden death has not been reliably proven. There are also very limited data regarding the pharmacological treatment of QT BCI, mainly related to the treatment of SQT1. One drug that shows great promise is hydroquinone, which has been shown to consistently prolong the QT interval and reduce episodes of ventricular tachycardia [12].
Conclusion Prolongation and shortening of the QT interval is often encountered in clinical practice and can cause sudden death in patients. Timely diagnosis allows you to choose the optimal treatment tactics and truly save the lives of such patients. Therefore, knowledge of methods for diagnosing and treating long and short QT interval syndromes is necessary for doctors of all specialties in their daily work.
Literature 1. Shkolnikova M.A. Primary, hereditary long QT syndrome // Long QT syndrome / Ed. M.A. Shkolnikova. M.: Medpraktika, 2001. P. 9–45. 2. Gollob MH, Redpath CJ, Roberts JD The Short QT Syndrome: Proposed Diagnostic Criteria // J. Am. Coll. Cardiol. 2011. Vol. 57. P. 802–812. 3. Nachimuthu S., Assar MD, Schussler JM Drug-induced QT Interval Prolongation // Ther. Adv. in Drug Safe. 2012. Vol.3(5). P.241–253. 4. Rautaharju PM, Surawicz B., Gettes LS AHA/ACCF/HRS Recommendations for the Standardization and Interpretation of the Electrocardiogram: Part IV: The ST Segment, T and U Waves, and the QT Interval: A Scientific Statement From the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society: Endorsed by the International Society for Computerized Electrocardiology // Circulation. 2009. Vol. 119. P.e241–e250. 5. Goldenberg I., Moss AJ, Zareba W. QT Interval: How to Measure It and What Is “Normal” // J. Cardiovasc. Electrophysiol. Vol.17. P. 333–336. 6. Viskin S. The QT interval: Too long, too short or just right // Heart Rhythm. 2009. Vol. 6. No.5. P. 711–715. 7. Schwartz PJ et al. Diagnostic criteria for the long QT syndrome. An update // Circulation. 1993. Vol. 88. P. 782–784. 8. Khan LA Long QT Syndrome: Diagnosis and Management // Amer. Heart J. 2002. Vol.143(1) 9. Roden DM Long-QT Syndrome // N. Engl. J. Med. 2008. Vol. 358. P.169–176. 10. Roden DM, Viswanathan PC Genetics of acquired long QT syndrome // J. Clin. Invest. 2005. Vol. 115. P. 2025–2032. 11. Morita H., Wu J., Zipes DP The QT syndromes: long and short // Lancet. 2008. Vol. 372. P. 750–763. 12. Patel C., Yan G.-X., Antzelevitch C. Short QT Syndrome: From Bench to Bedside // Circ. Arrhythm. Electrophysiol. 2010. Vol.3. P.401–408. 13. Bjerregaard P., Nallapaneni H., Gussak I. Short QT interval in clinical practice // Journal of Electrocardiology. 2010. Vol. 43. P. 390–395. 14. The Cardiac Society of Australia and New Zealand (CSANZ). Guidelines for the diagnosis and management of Familial Long QT Syndrome 2011. https://www.csanz.edu.au/documents/guidelines/clinical_practice/Familial_Long_QT_Syndrome.pdf
Types of compensatory pauses
There are two types of compensatory pauses:
- Full.
- Incomplete.
A complete compensatory pause after ventricular extrasystoles appears as a consequence of the fact that the passage of an extraordinary impulse through the atrioventricular node is not observed. The charge of the sinus node is not destroyed.
The next sinus impulse reaches the ventricles at the time when an extraordinary contraction occurs in them. This period is called the refractory period. The ventricles respond only to the next sinus impulse, which is equal in time to two cardiac cycles.
This means that the time indicating the intervals before and after the extrasystoles is equal to two normal intervals R - R.
An incomplete compensatory pause is characterized by the appearance of excitation in the ectopic focus. The impulse reaches the retrograde sinus node, after which the charge formed in it is destroyed. At this moment, another normal sinus impulse is formed. This means that the interval that appears after the extrasystole is equal to one normal R-R interval and the time during which the extrasystolic impulse travels from the ectopic focus to the sinus node. That is, this situation suggests that the distance from the sinus node to the ectopic focus affects the pause after the extrasystole.
The location of the ectopic focus and the atrioventricular node affects the interval of atrial extrasystole P – Q. The location of the node near the focus significantly shortens P – Q.
Classification
Sinus
Atrial
- atrial
- inferior atrial
- left atrial
From AV connection
- with simultaneous excitation of the atria and ventricles
- with previous excitation of the atria and ventricles:
- retrograde blockade 1st degree
- with complete retrograde block
- with complete anterograde blockade
Ventricular
- septal
- parietal
How does such a phenomenon threaten human health?
A compensatory pause is a cause for concern, and its occurrence always negatively affects the pumping function of the heart. This condition can appear after emotional excitement, drinking a lot of coffee, nicotine abuse, or sleep disturbances.
Of particular danger are compensatory pauses resulting from signals in the area of ischemic and infarct zones. Such cases, judging by statistical data, often lead to the development of spontaneous ventricular fibrillation, which, in turn, ends in the death of the patient.
A compensatory pause may be evidence of serious illnesses:
- heart defect,
- myocarditis,
- ischemic disease,
- myocardial infarction,
- arterial hypertension,
- chronic heart failure.
Treatment
In order to get rid of compensatory pauses, it is important to cure the underlying disease that provoked them. For this purpose, beta-blockers, sedatives and tranquilizers are used, with the help of which extrasystoles are reduced. Drugs based on quinidine cope well with arrhythmia.
In addition, sometimes it is necessary to seek the help of a psychotherapist.