Introduction
One of the rare and difficult to diagnose, predominantly congenital, primary cardiomyopathies is non-compact, or spongy, myocardium of the left ventricle, which is manifested by hypertrophy of the left ventricular myocardium, its excessive trabeculation and the formation of wide intertrabecular spaces due to disruption of the intrauterine process of myocardial compaction. Diagnosis of non-compact myocardium of the left ventricle is difficult due to the lack of typical clinical signs of the disease and lack of knowledge.
History of disease detection
The first information about non-compact myocardium of the left ventricle dates back to 1932, when S. Bellet, during an autopsy of a newborn with aortic atresia and coronary ventricular fistula, revealed a spongy structure of the myocardium. In the 60–70s of the last century, publications began to appear in foreign literature that described changes in the myocardium in the form of increased trabecularity in combination with various heart pathologies: obstruction of the outflow tract of the left and right ventricles, combined cyanotic congenital heart disease, atrial septal defect, interventricular septum, pulmonary stenosis, anomaly of the coronary arteries. However, this pathology is defined as non-isolated non-compact ventricular myocardium.
It was later shown that non-compact left ventricular myocardium can also occur in combination with neuromuscular diseases such as metabolic myopathy, Barth syndrome, Ohtahara syndrome and others. The literature describes cases of non-compact myocardium in hereditary defects of the facial skull - protruding forehead, bilateral strabismus, micrognathia, cleft palate, etc.
In 1984, an isolated pathology in the form of a sinusoid in the left ventricle was described. A year later, the authors described a case of diagnosed atypical dilated cardiomyopathy in a 21-year-old patient who was diagnosed with acute left ventricular failure at the age of 15 years. Echocardiographic examination revealed channel-like structures in the thickened and hypokinetic left ventricular myocardium; angiography revealed a “honeycomb” structure of the left ventricular wall. A year later, the same authors described a similar clinical case in a 21-year-old student during an autopsy with pronounced sinusoids in the hypertrophied myocardium. In 1990, the results of observation of 8 cases of non-compact myocardium in children were presented. Despite the fact that the number of publications on cases of non-compact myocardium of the left ventricle in the available literature is increasing, there is not enough information about isolated non-compact myocardium in children [1, 5, 6, 8, 10].
Epidemiology
The prevalence of non-compact left ventricular myocardium is, according to some authors, 0.05–0.24%, others – 0.014–0.14% in the general population. It can be assumed that due to the advent of the latest modern diagnostic methods, the detectability, and therefore the prevalence, of this pathology may increase. It is known that non-compact myocardium is more common in male patients than in female patients, ranging from 56 to 82% [8, 9].
The authors provide a link to data from 2003 on an epidemiological study of primary cardiomyopathies in Australian children, where non-compact myocardium was identified in 9.2% of cases of the total number of primary cardiomyopathies and was in third place after dilated and hypertrophic cardiomyopathies [1].
Pathogenesis
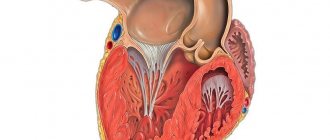
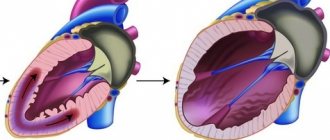
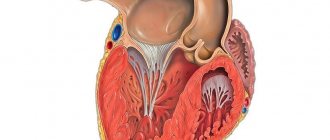
Disturbances in embryogenesis occur in the early stages. Normally, by the 26th day of intrauterine development, the myocardium is represented by a complex sponge-like structure of muscle trabeculae with multiple intertrabecular pockets - lacunae. This structure is a necessary condition for normal development, since in this period the coronary vessels have not yet been formed and young cardiomyocytes are forced to consume oxygen directly from the chambers of the heart. By the 31st day, the walls of the heart become denser, the lacunae close and partially participate in the formation of coronary vessels. If compaction of myocardial fibers does not occur for any reason, then the child is born with a rare anomaly - non-compact (spongy) myocardium. The main symptom of this disease is deep trabeculae in the myocardium of the left ventricle and the interventricular septum, which entails a decrease in systolic function of the left ventricle. Complete non-compaction of the myocardium occurs in children and is usually combined with other congenital anomalies, most often stenoses of the outflow tract of the right and left ventricles. The mortality rate among children with concomitant pathology of complete non-compact myocardium is very high. Adults with isolated spongy myocardium usually have no complaints; this pathology can be detected accidentally during examination due to other conditions.
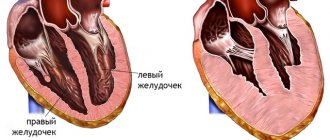
It has been established that in a healthy child the left ventricle has up to three visible trabeculae and is less trabecular compared to the right ventricle. In the case of intrauterine disturbance of endomyocardial morphogenesis, processes of disorganization of myocardial compaction occur. During embryogenesis, the depressions interact with the ventricular endocardium and, with further compaction of the myocardium, are transformed into capillaries. Non-compact myocardium is represented by the presence of more than three deep intertrabecular depressions in hypertrophied segments of the left ventricular myocardium. In addition, the ventricles are often hypokinetic. It was found that the deep intertrabecular recesses are covered with epithelium, which indicates a relationship with the ventricular endocardium. In contrast to established disorders in the left ventricle, it is not possible to differentiate increased trabecularity of the right ventricle, since the right ventricle normally has increased trabecularity. However, the etiopathogenetic mechanisms of the development of the disease have not yet been fully studied [1, 2, 9].
Isolated spongy myocardium
Disturbances in embryogenesis occur in the early stages. Normally, by the 26th day of intrauterine development, the myocardium is represented by a complex sponge-like structure of muscle trabeculae with multiple intertrabecular pockets - lacunae. This structure is a necessary condition for normal development, since in this period the coronary vessels have not yet been formed, and young cardiomyocytes are forced to consume oxygen directly from the chambers of the heart.
By the 31st day, the walls of the heart become denser, the lacunae close and partially participate in the formation of coronary vessels. If compaction of myocardial fibers does not occur for any reason, a child is born with a rare anomaly - non-compact (spongy) myocardium. Complete non-compaction of the myocardium occurs in pediatric practice and, as a rule, is combined with other congenital anomalies. Most often these are stenoses of the outflow tract of the right and left ventricles. The mortality rate among such patients is very high. In adult patients, isolated spongy myocardium may not be accompanied by clinical symptoms and may be detected during examinations for another reason. Morphologically, the non-compact myocardium is represented by a large number of hypertrophied trabeculae with intertrabecular pockets. The scarring is caused by deep invaginations lined by endothelium and penetrating close to the epicardial surface. The pockets have a direct connection with the cavity of the left ventricle, but there is no connection with the coronary circulation.
This myocardial structure is observed in some vertebrates - fish, reptiles, amphibians. Most researchers agree that echocardiography is the leading method in determining this pathology. Moreover, it is believed that the specific changes identified by this method are sufficient to make a diagnosis. Echocardiographic signs of spongy myocardium are: absence of other cardiac abnormalities; thickening of the left ventricular myocardium with increased trabecularization of more than one segment of the left ventricular wall, two-layer myocardium, the thickness of the spongy layer is 2 times greater than the thickness of the unaffected myocardium; extensive intertrabecular pockets communicating with the cavity of the left ventricle.
For a long time, a patient with isolated spongy myocardium may not have any complaints. Family cases of the disease are described, and there are references to sudden death or death in early childhood among relatives. Ventricular arrhythmias and embolic complications are common. The causes and electrophysiological mechanisms are still unknown. It has been suggested that the disordered structure of myocardial trabeculae, isometric contraction with increased intramural tension, and limitation of coronary blood supply in areas with non-compact myocardium may be prerequisites for life-threatening arrhythmias.
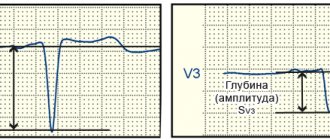
With this anomaly, there were signs of myocardial ischemia with changes in the final part of the ventricular complex on the electrocardiogram. Angiography in such patients reveals coronary arteries, and autopsy reveals ischemic changes in trabeculae and compacted myocardium.
A number of researchers have assessed regional myocardial blood flow using positron emission tomography. It has been suggested that the cause of ischemia is dysfunction of the myocardial microvasculature. Moreover, the predominant location of necrosis and fibrosis was the subendocardial non-compact layer, rather than the epicardial compacted one. When exposed to dipyridamole, a deterioration in blood supply was found in both the non-compacted and compacted layers, with the former being significantly greater.
Genetic aspects
The literature contains data on both sporadic forms of the disease and familial cases of isolated non-compact myocardium of the left ventricle. Isolated non-compact left ventricular myocardium is a genetically heterogeneous disease. The authors distinguish several types of left ventricular non-compact myocardium syndrome: type 1 - thickening of the left ventricular wall and interventricular septum without left ventricular dilatation is observed, type 2 - a combination of signs of left ventricular non-compact myocardium and dilated cardiomyopathy is observed, and X-linked type.
Type 1 syndrome of non-compact left ventricular myocardium is inherited in an autosomal dominant manner, in some cases it is the result of new mutations. In this type, both isolated syndrome of non-compact left ventricular myocardium and its combination with heart defects are observed. The most common defects are atrial septal defects, isolated or multiple ventricular septal defects, pulmonary artery stenosis and other defects. The cause of this disease is mutations in the dystrobrevin gene, which is located at the 18q12.1-q12.2 locus.
Type 2 is inherited in an autosomal dominant manner. There is no combination of left ventricular non-compact myocardium syndrome with heart defects. The gene responsible for this disease is still not known, but the 11p15 locus linked to this disease has been mapped. Therefore, direct diagnosis of this disease is not yet possible.
The X-linked type is inherited as an X-linked recessive type. This rare hereditary disease develops as a result of mutations in the G 4.5 (TAZ) gene, which is located on the X chromosome in the Xq28 region. This gene encodes the taffazin protein, which is an essential structural component of the membranes of skeletal and cardiac muscles, and also takes part in myocardial morphogenesis. Barth syndrome, other X-linked infantile cardiomyopathies, and X-linked endocardial fibroelastosis are associated with this gene. Although this mutation was not detected in adult patients, which may indicate other causes of the disease. Direct DNA diagnosis of this type of disease can confirm the presence of the disease. Women can be carriers of a mutant gene; they have a 50% chance of passing on the gene carrying the mutation to their sons. All daughters of female carriers will be healthy, but half of them will also be carriers of the mutant gene. However, not every mother who gives birth to a boy with left ventricular non-compact myocardium syndrome is diagnosed with a mutant gene. This fact can be explained by the fact that the mutation occurred for the first time in the patient. To determine the carriage of a mutant gene, molecular genetic examination methods are necessary [7].
Non-compact left ventricular syndrome
Left ventricular non-compaction syndrome ( spongy myocardium ) is one of the rare types of cardiomyopathies. in accordance with the definition and classification of cardiomyopathies by WHO (1995), it belongs to the category of “unclassified cardiomyopathies”. The disease is a disorder of myocardial development during embryonic development. The main symptom of this disease is deep trabeculae in the myocardium of the left ventricle and the interventricular septum, which entails a decrease in the systolic function of the left ventricle. In some cases, the pathological process may involve the myocardium of the right ventricle. The disease is clinically and genetically heterogeneous. As a rule, with this disease there is thickening of the walls of the interventricular septum and the left ventricle without its dilatation. The combination of dilated cardiomyopathy and signs of non-compact left ventricular myocardium may indicate one of the types of dilated cardiomyopathy (DCM, 1C, MIM: 601493), caused by mutations in the LDB3 , which is located in the 10q22.2-q23.3 region. DNA diagnostic capabilities: Currently, the search for mutations in the LDB3 in CMG is not carried out, but it can be developed upon request. LVM syndrome type 1 (MIM: #604169) is inherited in an autosomal dominant manner, with some cases resulting from new mutations. In this type, both isolated LVNC syndrome and its combination with heart defects are observed. The most common types of defects are atrial septal defects (ASD), isolated or multiple ventricular septal defects (VSD), and pulmonary artery stenosis, but some other types of defects have been described. The cause of this disease is mutations in the dystrobrevin gene (DTNA), which is located at the 18q12.1-q12.2 locus. DNA diagnostic capabilities: Currently, the search for mutations in the DTNA gene in CMG is not carried out, but it can be developed upon request. LVM syndrome type 2 (MIM: #604169) is inherited in an autosomal dominant manner. In this type, as a rule, there is no combination of LVNC with heart defects. The gene responsible for this disease is currently unknown, but the 11p15 locus has been mapped to be associated with this disease. Possibilities of DNA diagnostics: At present, direct DNA diagnostics of this disease is impossible. LVM syndrome, X-linked type (MIM: #300183) is inherited in an X-linked recessive manner. This is a rare hereditary disease that develops as a result of mutations in the G4.5 ( TAZ ) gene located on the X chromosome in the Xq28 region. This gene encodes the protein tafazzin, which is an essential structural component of the membranes of skeletal and cardiac muscles and takes part in myocardial morphogenesis. Possibilities of DNA diagnostics: Since 2006, CMG has been conducting direct DNA diagnostics of this disease. Incidence The incidence of left ventricular non-compaction syndrome (LVSC) is unknown. This disease is believed to be rare. Inheritance: LVNC is inherited in an X-linked recessive manner. Women can be carriers of a mutant gene, and they have a 50% chance of passing on the gene carrying the mutation to their sons. All daughters of female carriers will be healthy, but half of them will also be carriers of the mutant gene. It is important to remember that not every mother who gave birth to a boy with LVNC syndrome is a carrier of the mutant gene. Some cases of this disease are the result of mutations that first appeared in the patient. The question of whether a woman is or is not a carrier of the disease can only be resolved using molecular genetic methods. Clinical symptoms: The main symptom of this disease is deep trabeculae in the myocardium of the left ventricle of the heart and/or the interventricular septum. Most often, the pathological process is localized in the region of the apex of the heart, the lower and lateral walls of the left ventricle. Some patients may exhibit some symptoms of Barth syndrome (there should be a link to the page with Barth syndrome here). Some patients develop ventricular rhythm and conduction disturbances. Cases of the formation of blood clots in the trabecular area and thromboembolic complications have been described. Treatment: There is currently no specific treatment for LVNC syndrome. In some cases, surgical treatment is possible. The main threat to the health and life of patients is the development and progression of heart failure. Prevention of thromboembolic complications is necessary. Diagnosis: To diagnose LVNC syndrome it is necessary: - Ultrasound examination of the heart; — ECG and Holter monitoring; — search for mutations in the TAZ gene using molecular genetic methods in patients, as well as diagnosis of heterozygous carriage in relatives of patients (the study can be performed at the CMG). It is important to know that medical genetic counseling is recommended for families in which cases of LVNC syndrome have been observed.
When conducting prenatal (antenatal) DNA diagnostics in relation to a specific disease, it makes sense to diagnose common aneuploidies (Down, Edwards, Shereshevsky-Turner syndromes, etc.) using existing fetal material, paragraph 54.1. The relevance of this study is due to the high total frequency of aneuploidy - about 1 in 300 newborns, and the absence of the need for repeated sampling of fetal material.
Non-compact left ventricular syndrome
Classification
Since 1995, at the proposal of the WHO, according to the classification of cardiomyopathies, non-compact myocardium has been classified as a group of unclassified cardiomyopathies. In accordance with the classification of cardiomyopathies proposed by the American Heart Association (AHA, 2006), non-compact left ventricular myocardium belongs to primary genetic cardiomyopathies (unclassified cardiomyopathies). Since 2008, the European Society of Cardiology has proposed a classification of known and rare cardiomyopathies. In accordance with this classification, two large groups of cardiomyopathies are distinguished: familial and non-familial. The familial form is diagnosed when the disease is present in more than one family member or when the same genetic mutation occurs in cases of diseases in the family. The familial form of cardiomyopathy is often a monogenic disease. Of the congenital nonfamilial cardiomyopathies, only idiopathic ones were included in the classification. Acquired non-familial forms of cardiomyopathies are not included in this classification, since acquired cardiomyopathies have significant differences in diagnosis and treatment due to the fact that they are a complication of the disease, and not its clinical sign [3, 7–9].
Clinical signs
The main symptoms of non-compact myocardium are manifestations of heart failure, arrhythmias (ventricular tachycardia, atrial fibrillation, Wolff-Parkinson-White syndrome), thromboembolism (pulmonary embolism, blood clots in the ventricles of the heart), especially pronounced in patients with reduced left ventricular function. In addition to cardiac symptoms, facial dysmorphism was often observed in children. In adults, complete block of the bundle branches is most often observed. Also, children were more likely to have a familial type of the disease than adults. A constant sign of the disease in children and adults was the presence of non-compact myocardial segments. The severity of the condition of patients of both age groups was determined by the degree of myocardial non-compaction [1–3, 8, 9].
Diagnostics
To diagnose non-compact myocardium of the left ventricle, various methods of instrumental examination are used: echocardiography, magnetic resonance imaging, less often - computed tomography, positron emission tomography.
To visualize the cavity of the left ventricle, echocardiography is used, often two-dimensional, as well as three-dimensional. It is especially important to use echocardiography when conducting family screening in order to identify patients in the absence of complaints and symptoms of the disease.
The main echocardiographic sign of non-compact myocardium is the presence of increased trabecularity of the left ventricle. However, increased trabecularity may be a normal variant, but with non-compact myocardium, it can most often be determined between the anterolateral wall and the interventricular septum. For a more accurate diagnosis of non-compact myocardium, several echocardiographic studies are recommended, since the existing pathological trabecularity can be visually smoothed out when obtaining flat sections. It is also recommended to use parasternal short-axis imaging at end systole for differential diagnosis.
There are several options for diagnostic criteria for isolated non-compact left ventricular myocardium.
Criteria for diagnosing isolated non-compact myocardium of the left ventricle according to TK Chin (1990): absence of any other concomitant structural abnormalities of the heart; parasternal long axis, subcostal and apical views; determining the depth of excavations; measured at the end of diastole, the ratio between the distances from the surface of the epicardium to the beginning of the notches and from the epicardium to the end of the trabeculae is ≤ 0.5; increased trabecularity and deep intertrabecular depressions.
Criteria for the diagnosis of isolated non-compact myocardium of the left ventricle according to R. Jenni (1999): absence of concomitant cardiac anomalies; visualization at the level of the parasternal short-axis and apical projection reveals a typical two-layer myocardial structure with a thin compact outer (epicardial) layer and a thicker non-compact inner layer (endocardial) layer, which consists of a trabecular mesh structure with recesses deep to the endocardium (the maximum end-systolic ratio of the non-compact endocardial layer and compact myocardium is 2); segmental localization of abnormal myocardium predominates (that is, non-compact myocardium dominates at the apex (about 80%) and in the middle sections of the lower and lateral walls of the ventricle); using color Doppler, deep perfusion of intertrabecular grooves can be detected (unlike myocardial sinusoids, intertrabecular grooves do not communicate with the coronary blood flow).
In 2006, S. Lilje and co-authors proposed a method for quantitatively assessing the degree of myocardial non-compaction depending on the values of the above ratio: a ratio value of 0.33–0.26 corresponds to “mild” non-compaction, 0.25–0.2 – moderate, less than 0 ,2 - heavy. This value is recognized as a prognostic sign of the further development of the disease and the occurrence of complications, since it closely correlates with the degree and rate of development of heart failure.
Authors C. Stollbeger and J. Finsterer believe that to make a diagnosis of non-compact myocardium of the left ventricle, the following conditions are necessary: the presence of more than three trabeculae extending from the wall of the left ventricle in the apex to the papillary muscle and visualized in at least one position; perfusion of the intertrabecular spaces of their ventricular cavity, recorded by Doppler echocardiography.
The above criteria for diagnosing isolated non-compact myocardium of the left ventricle are echocardiographic signs confirming the diagnosis.
It has been established that most often (about 80%) the apical and middle segments of the lower and lateral walls of the left ventricle are affected.
The magnetic resonance imaging method for diagnosing isolated non-compact myocardium of the left ventricle is the most informative, especially when using three-dimensional visualization. The computed tomography method is less commonly used. Both of these methods have high resolution and are considered very promising in the diagnosis of non-compact myocardium of the left ventricle, although the results of their use are not sufficiently described in the literature [4–6, 8, 10].
Cardiomyopathy - symptoms and treatment
According to modern concepts, the patient's treatment strategy is determined by dividing patients into categories depending on the type of cardiomyopathy.
All patients with detected cardiomyopathy, regardless of the course of the disease (including asymptomatic ones), require dynamic monitoring . The frequency of observation and the volume of examinations are determined individually. The mandatory list includes standard tests (clinical and biochemical blood tests), ECG, echocardiography and Holter ECG monitoring.
Treatment tactics depend on many factors and are selected individually. This takes into account anatomical features - obstruction of the left ventricular outflow tract, distension of the heart cavities, the presence of valvular pathology, the stage of heart failure and also concomitant diseases. It is necessary to identify factors that increase the risk of sudden death and life-threatening arrhythmias [15]
General measures include limiting significant physical activity and avoiding sports that may cause further stress on the myocardium [9]. But patients with cardiomyopathy do not require complete exclusion of physical activity and bed rest [10]. The level of loads, their frequency, intensity and duration are selected individually. It is recommended to avoid drinking alcohol and smoking.
With dilated cardiomyopathy, it is necessary to treat the cause of the development of stretching of the heart cavities, if possible. All standard groups of drugs are used in drug therapy for heart failure:
- ACE inhibitors;
- angiotensin II receptor blockers;
- beta blockers;
- aldosterone receptor blockers;
- diuretics;
- digoxin.
For the treatment of severe heart failure, combination drugs containing sacubitril and valsartan, as well as an implantable cardioverter-defibrillator and/or cardiac resynchronization therapy are recommended. Oral anticoagulants are used in patients with cardiac arrhythmias [12].
In the treatment of obstructive cardiomyopathy, some groups of drugs have limitations (ACE inhibitors, angiotensin II receptor blockers), but beta-blockers and calcium channel blockers are used. If necessary, antiarrhythmic drugs are used [13].
Treatment of restrictive cardiomyopathy focuses on treating the underlying disease causing the changes in the heart. The use of diuretics is possible [14].
In addition to medications, in some cases they resort to surgical methods for treating cardiomyopathy .
For hypertrophic cardiomyopathy, septal myectomy is used - excision of the myocardium located at the base of the interventricular septum. It can be supplemented by intervention on the altered mitral valve: valvuloplasty, mitral valve replacement and correction of the mitral valve ring.
In case of severe obstructive hypertrophic cardiomyopathy, surgery is considered - excision of part of the heart muscle or a more gentle technique - percutaneous transluminal alcohol ablation . In this case, up to 3 ml of 96% alcohol is injected through a catheter into the zone of maximum myocardial hypertrophy and its infarction is caused. Because of this, the muscle decreases in size, and the obstruction (obstruction) to blood flow through the mitral ring disappears. Next, a pacemaker is installed to synchronize the work of all parts of the heart. This procedure is performed by cardiac surgeons in specialized departments.
To prevent arrhythmia, some patients are given a special type of heart stimulator - a defibrillator-cardioverter, which ultimately prolongs their life [2].
And of course, in especially difficult situations, the possibility of heart transplantation is considered to save lives. These are unique operations carried out in specialized centers both in the Russian Federation and abroad. Less than 100 such operations are performed annually in the Russian Federation. A heart transplant patient requires lifelong monitoring at a transplant center and takes a number of powerful drugs that affect the immune system [11].
Treatment
The basis of therapeutic tactics in patients with spongy myocardium is the prevention and treatment of heart failure, heart rhythm disturbances and thromboembolic complications (taking anticoagulants for impaired left ventricular function). Asymptomatic patients and patients without arrhythmias and signs of left ventricular dysfunction are not prescribed treatment. Some patients are indicated for heart transplantation. Patients with malignant rhythm disorders are recommended to have a cardioverter-defibrillator implanted [1, 2, 7, 9].
The prognosis for patients with non-compact myocardium of the left ventricle depends on the volume of the affected segments, the overall contractility of the myocardium, the time of occurrence and the rate of increase of symptoms of heart failure. According to I. Jedlinsky and co-authors, mortality within 6 years was 50%. Of the 34 patients observed in the study by E. Oechslin et al., 12 patients died over a 44-month period, 6 of whom were diagnosed with sudden cardiac death, 4 with terminal heart failure, four patients underwent a heart transplant, and another four received a cardioverter-defibrillator. . The prognosis is especially poor in patients with an ejection fraction of less than 35%. Factors indicating an unfavorable prognosis of the disease include: large end-diastolic size of the left ventricle, persistent atrial fibrillation, and bundle branch block on the electrocardiogram.
During pathological studies , it was established that isolated non-compact myocardium is characterized by the presence of hypertrophied segments of the left ventricular wall, which consists of a non-compact endocardial layer, in which increased trabecularity is noted, and a thin epicardial compact layer. Deep intertrabecular recesses communicate with the cavity of the left ventricle, but not with the coronary blood flow, in contrast to non-compact myocardium associated with other congenital heart defects, when intertrabecular recesses communicate with the cavity of the left ventricle and the vessels of the coronary circulation (non-insulated myocardium) [1, 2, 8 , 10].
Pathogenesis and clinic
The pathogenesis consists of heart failure, arrhythmia syndrome, and thromboembolic syndrome.
Heart failure takes a leading place in the clinical picture of this disease. Due to the disturbed architectonics of the myocardial structure, its contractility is impaired. With a severe degree of non-compactness, the clinical picture of this syndrome is most pronounced with a predominance of insufficiency in both circulation circles and with a pronounced decrease in the overall contractility of the myocardium. As a rule, in some patients the clinical picture of the disease can manifest itself as a result of the initiation of an inflammatory process in the myocardium, and in some cases, with a favorable course, non-compact myocardium can be detected for the first time in elderly people. In the clinical picture of the disease, pain, which is expressed by attacks of angina, may come first. This is explained by the greater need for oxygen in the non-compact layer of the myocardium, which is not supplied with blood by the main branches of the coronary arteries, and its blood supply occurs directly from the cavity of the left ventricle.
The syndrome of rhythm disturbances is most often manifested by ventricular rhythm disturbances - in more than half of the cases (extrasystole, often of high gradations). In a quarter of cases, atrial fibrillation is observed. Also, due to endomyocardial fibrosis with the capture of the conduction system of the heart, conduction disturbances of the type AB and SA blockades are observed.
Of course, a violation of global systolic function, the presence of cardiac arrhythmias, and in particular atrial fibrillation, the presence of deep lacunae predisposes to the formation of blood clots in the cavity of the left ventricle, and hence thromboembolic complications (cardioembolic strokes, TIA, mesenteric thrombosis).
Clinical case of non-compact myocardium of the left ventricle in a 6-year-old girl
The child Melania B. was born from the first pregnancy, which occurred against the background of chronic maternal pyelonephritis and chronic intrauterine fetal hypoxia, from the first urgent birth at 39 weeks of gestation with a body weight of 3700 g. A short umbilical cord was noted during childbirth. On the 1st day after birth, a birth injury was diagnosed - rotational subluxation of the first cervical vertebra C1, which was clinically manifested by signs of movement disorder syndromes and vegetovisceral disorders. Neurosonography revealed ultrasound signs of grade I–II periventricular ischemia. Due to the presence of signs of post-hypoxic syndrome of maladjustment of the cardiovascular system, the girl underwent echocardiography, which revealed echo signs of a minor anomaly of heart development - an abnormal chord of the left ventricle, hypertrabecularity of the left and right ventricles, proliferation of papillary muscles, carditis.
From the maternity hospital, the girl was transferred to the neonatal pathology department. In addition to the listed examinations, echocardiography is provided on the 12th day of life: the heart chambers are not dilated, no pathological shunts are identified, chordal features of the left ventricle, systolic-diastolic function of the left ventricle is not changed. The electrocardiogram shows slowing of atrioventricular conduction, diffuse changes in the ventricular myocardium. After treatment, the girl was discharged home at the age of 1 month with recommendations for observation by a cardiologist, neurologist, and pediatrician.
Subsequently, the child’s condition gradually worsened, symptoms of heart failure increased, and therefore the girl at the age of 5 months was hospitalized in the cardio-rheumatology department of the Lugansk city multidisciplinary children's hospital No. 1 with a diagnosis of congenital non-rheumatic carditis of severe degree, decompensated, circulatory disorder of degree IIA, thymomegaly II degree, minor cardiac anomaly, motor impairment syndrome, perinatally caused. When examined on an electrocardiogram, there is dilatation of the left ventricle with systolic overload, diffuse changes in the myocardium. A repeat electrocardiogram 4 days later shows dilatation of the left ventricle and left atrium with secondary mitral insufficiency of degree IIB, possibly due to previous myocarditis, and a decrease in the systolic and diastolic function of the left ventricle. An echocardiogram after 9 days showed moderate dilation of the left chambers of the heart, decreased contractility of the left ventricular myocardium, and moderate individual increased trabecularity of the left ventricle. Consultation with a cardiac surgeon: there is no heart defect. In the department, the child was prescribed digoxin for the first time. The girl was discharged from the department at the insistence of her parents at the age of 5 months 13 days.
At the age of 8 months, the girl’s condition worsened; she was again hospitalized in the cardio-rheumatology department with a diagnosis of congenital non-rheumatic carditis, severe. Stage of decompensation, circulatory disorder IIB degree, relative insufficiency of the mitral and tricuspid valves, slowdown of statokinetic function, rickets II degree, subacute course, malnutrition I degree, immunodeficiency state. Two weeks later the girl was discharged home with improved condition.
Subsequently, the child was repeatedly consulted by a cardiologist, examined, and received treatment. An electrocardiogram at the age of 9 months revealed signs of incomplete blockade of the right bundle branch.
At the age of 1 year 3 months, the girl was examined and consulted in the scientific and practical medical center of pediatric cardiology and cardiac surgery in Kiev, where the diagnosis was made: cardiomyopathy, non-compact myocardium of the left ventricle, severe mitral regurgitation, high pulmonary hypertension. Recommendations were given for further tactics of examination and treatment of the patient. Subsequently, the girl was regularly examined and treated at the specified center.
Cardiac magnetic resonance imaging without intravenous contrast was performed at 3 years and 2 months of age. A series of tomograms show enlargement of the cavities of both ventricles (the size of the left ventricle is 5.6 x 3.6 x 3.6 cm, the right ventricle is 4.4 x 2.25 cm), the left atrium is significantly enlarged, and moderate insufficiency of the atrioventricular valves is observed. The endocardium of the left ventricle has an open trabecular structure with a relative thinning of the compact subepicardial part up to 0.25 cm. The diastolic ratio of the non-compact and compact parts of the left ventricle is 1.25/0.25 cm. Similar structural changes in the myocardium are determined in the right ventricle. Left ventricular ejection fraction - 27%. A small amount of fluid in the pericardial cavity. The girl has not been genetically examined.
Due to deterioration of her condition, the girl was again referred for treatment at the age of 5 years and 3 months. Diagnosis: non-compact myocardium of the left ventricle. Severe tricuspid and mitral valve insufficiency, dilatation of the left heart, severe pulmonary hypertension. An echocardiogram shows the left chambers of the heart are dilated, myocardial contractility is satisfactory. Pronounced trabecularity of the posterior wall of the left ventricle, the thickness of the compact part is 2.5 mm, the non-compact part is 12 mm. Conclusion: non-compact myocardium of the left ventricle, insufficiency of the tricuspid and mitral valves, dilatation of the left chambers of the heart, severe pulmonary hypertension. Repeated echocardiography after 10 days of treatment revealed: the left chambers of the heart are enlarged, myocardial contractility is good (positive dynamics during treatment). Diastolic disorders of the left ventricle type III (restrictive). Severe trabecularity of the left ventricle (non-compact myocardium). There is no fluid in the pericardial cavity and pleural cavities. During the treatment period, the girl received dobutamine, Lasix, asparkam, digoxin, berlipril, panangin, and veroshpiron. Considering the positive dynamics of the patient’s clinical condition, the council of doctors decided that surgical treatment was not indicated and it was recommended to continue treatment on an outpatient basis. Currently, the girl's condition is compensated.
Clinical and instrumental characteristics of non-compact myocardium of the left ventricle
Ultrasound machine HM70A
Expert class at an affordable price.
Monocrystal sensors, full-screen display mode, elastography, 3D/4D in a laptop case. Flexible transformation into a stationary scanner with a cart.
Introduction
Non-compact left ventricular myocardium (LVCM) is a genetically determined myocardial lesion manifesting as heart failure (HF), arrhythmias, thromboembolism and sudden cardiac death. Due to its rarity and the absence of specific complaints and physical findings, LVNC was first described as a nosological entity in 1984 by R. Eng berding and F. Bender [1]. According to echocardiographic studies in adults, LV NML is rarely detected - with an average frequency of 0.014%, but in patients with a decrease in ejection fraction
Etiology
LVNC is a hereditary disease with an autosomal dominant mode of inheritance. The first of the genes is localized on chromosome Xq28 and is related to mitochondrial function and regulation of cardiolipin levels. The second gene encodes α-dystrobrevin, a protein involved in the formation of the dystrophin-associated glycoprotein complex. The manifestation of changes in the heart in LVNC is observed already in the prenatal period. At the 5-8th week of embryonic development, the formation of dense myocardium from epic to endocardium and from the base of the heart to the apex is disrupted. At the same time, the coronary circulation is formed, the intertrabecular spaces are reduced to the size of capillary vessels. It is during this period that the basis of the non-compact myocardium is laid: 1 - a thick, non-compact spongy layer of the heart muscle, formed by trabeculae, having an underdeveloped vascular system; 2 - a thin layer of normal homogeneous myocardium, capable of normal contraction. The disease can manifest at different ages.
Based on morphology, there are 3 types of non-compact myocardium:
- Lacunar - non-compact myocardium is represented by a network of well-visualized trabeculae with wide, deep lacunae.
- Spongy - non-compact myocardium is represented by alternating many tiny lacunae (less than 1 mm) and trabeculae, difficult to distinguish from each other.
- Mixed - trabeculae and lacunar spaces are visualized, less pronounced than with the lacunar type.
Clinical manifestations of LVNC are nonspecific and include: shortness of breath, edema, thromboembolism (4-24%) and tachyarrhythmia (in 47% of cases, ventricular tachyarrhythmias occur (in 40% of patients this is the first manifestation of the disease)), supraventricular tachycardia (44%), atrial fibrillation (AF) (25%), WPW syndrome (15%). Due to the high incidence of severe rhythm disturbances—ventricular tachycardia—in such patients, sudden death may be the first manifestation of the disease.
Myocardial trabecularity promotes the formation of intraventricular thrombi, which explains the high incidence of thromboembolic complications (21-38%), clinically manifested by stroke, transient ischemic attacks, pulmonary embolism and thrombosis of mesenteric vessels.
Diagnostics
In 2001, R. Jenni et al. [2] proposed diagnostic criteria for the disease. The diagnosis is valid if all 4 criteria are present.
- Typical two-layer myocardial structure with a thin, compact outer (epicardial) layer and a significantly thickened non-compact inner (endocardial) layer (maximum end-systolic ratio of non-compact endocardial layer to compact myocardium >2 in adults and up to 1.4 in children).
- Documented blood flow in deep intertrabecular spaces according to color Doppler ultrasound (unlike myocardial sinusoids, intertrabecular spaces do not communicate with the coronary vessels).
- The dominant localization of non-compact myocardium (>80%) is in the apical zone and in the middle sections of the lateral and posterior wall of the LV.
- No concomitant cardiac anomalies.
C. Lilje et al. [3] in 2006 proposed quantitative criteria for determining the degree of non-compactness of the myocardium in relation to the layer of “dense” true myocardium (X) to the thickness of the entire heart wall at the level of the LV apex (Y): 0.33-0.26 - “soft” non-compactness; 0.25-0.2 - moderate; less than 0.2 - heavy.
This value closely correlates with the degree and speed of development of heart failure and has prognostic significance for the further development of the disease and the occurrence of complications.
Differential diagnosis
Studies report that many patients were misdiagnosed: in descending order of frequency, DCM, HCM, fibroelastosis, myocarditis, restrictive cardiomyopathy, pericarditis, and LV thrombosis.
Prognosis and predictors of unfavorable course of the disease. Mortality within 6 years is about 50%. In children, the mortality rate is about 17.1%. Predictors of poor prognosis:
- Increased LV end-diastolic size on initial echocardiography in a patient.
- Chronic heart failure class III-IV according to NYHA.
- Permanent form of AF.
- Bundle branch block.
The prognosis is also worsened by the presence of ventricular tachyarrhythmias, which is often the cause of sudden cardiac death in patients with non-compact myocardium, which may be the first and only manifestation of LVNC.
Treatment
There is no specific therapy for LVNC. Symptomatic treatment of congestive heart failure, arrhythmias and prevention of thromboembolic complications are carried out. In terminal stages, heart transplantation is possible.
Clinical observation
Patient G., born in 1962, was admitted urgently to the emergency department with complaints of severe shortness of breath, including at rest, orthopnea, abdominal enlargement, decreased amount of urine, swelling of the lower extremities, severe weakness with little physical activity. He considers himself sick over the last month, when a low-grade fever appeared, shortness of breath began to increase during physical exertion and in a horizontal position, swelling appeared in the legs, and the abdomen became enlarged. Took antibiotics for 5 days. Body temperature returned to normal, but health did not improve.
Upon examination, the general condition is serious. Consciousness is clear. The physique is correct. The food is satisfactory. The skin is pale. The sclera is subicteric. Visible mucous membranes are cyanotic. Massive swelling of the lower extremities. Pulse 102 beats/min, rhythmic, low filling. Blood pressure 140/80 mm Hg. Swelling of the neck veins. Cyanosis of the face in a horizontal position. The tones are dull, systolic murmur with a maximum at Botkin's point. Respiratory rate 19 per minute. The abdomen is enlarged due to ascites. On superficial palpation it is tense.
In the blood test, attention is drawn to a 24-fold increase in D-dimer. Ultrasound examination of the thoracic and abdominal organs: enlargement and diffuse changes in the liver. Enlargement and diffuse changes in the pancreas. Calicoectasia and cysts of the parenchyma of the left kidney. Free fluid in the abdominal cavity. Bilateral hydrothorax. Computed tomography: indirect signs of thromboembolism of small branches of the pulmonary artery. Signs of infarction pneumonia SX of the right and left lung.
According to the ECG, there was: sinus tachycardia with a frequency of 104 beats/min. Enlargement of the left and right atrium. Pronounced voltage criteria for LV hypertrophy. Secondary changes in the LV myocardium associated with LV hypertrophy. Incomplete blockade of the left bundle branch (QRS 110 ms).
An echocardiographic study performed on an EKO7 ultrasound system (Samsung Medison) revealed a significant dilatation of the aorta at the level of the sinuses of Valsalva and the ascending limb - 47/48 mm. Left atrium: anteroposterior size 50 mm. Long axis - 73 mm. LV end-diastolic dimension was 75 mm, LV end-systolic dimension was 63 mm, LV ejection fraction was 33%. Right ventricle (at the level of the tricuspid valve) - 47 mm. Right atrium - 47 x 70 mm. Significant expansion of the pulmonary artery (PA) trunk - 41 mm. The splitting of the pericardial layers behind the posterior wall of the LV is 5 mm, behind the right atrium - up to 6 mm. Mitral regurgitation degree II. Tricuspid regurgitation grade III. Aortic regurgitation degree II. Regurgitation on the pulmonary valve - II degree. The average hemodynamic pressure in the PA (based on tricuspid regurgitation) is 46 mm Hg. The maximum pressure in the LA is 72 mm Hg.
The LV wall in the middle and apical region consists of 2 layers: the outer, 5.5 mm thick, of a homogeneous structure (compact myocardium) and the inner, represented by wide trabeculae, lacunae, reaching the compact layer (like a spongy structure). Blood flow is recorded throughout the lacunae. The thickness of the non-compact layer in the middle part of the LV is up to 16 mm, in the apex region up to 29 mm. On the scanograms (Fig. 1-4), with a good level of ultrasound visualization, clear signs of non-compaction of the LV myocardium are visible. Thus, there is a typical echocardiographic picture of LVNC. This clinical and echocardiographic description represents the first clinical detection and description of LVNC in the Volga Federal District.
Rice. 1.
Five-chamber position when located from the top.
Rice. 2.
Transverse parasternal position.
Rice. 3.
Non-compact structure of the LV myocardium.
Rice. 4.
Non-compact structure of the LV myocardium.
conclusions
- Identification of specific echocardiographic signs of non-compaction allows an accurate diagnosis of LVNC.
- The lack of theoretical and practical skills among most practitioners in identifying LVNC is associated with low prevalence in the population and insufficient awareness (less than 20 years of detection experience).
- Knowledge of clinical diagnostic criteria for LVNC by cardiologists and ultrasound doctors will contribute to early diagnosis and treatment of patients with this disease.
- Targeted echocardiography screening of the patient's relatives is necessary for early diagnosis and treatment.
Literature
- Engberding R., Bender F. Identification of a rare congenital anomaly of the myocardium by twodimentional echocardiography: persistence of isolated myocardium sinysoids // Am J Cardiol. 1984; 53: 1733-1734.
- Jenni R., Oechslin E., Schneider J. et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular noncompaction: a step towards classification as a distinct cardiomyopathy // Heart. 2001; 86: 666-671.
- Lilje S., Razek V., James J. et al. Complications of non-compaction of the left ventricular myocardium in a pediatric population: a prospective study // Eur Heart J. 2006; 27 (15): 1855-1860.
Ultrasound machine HM70A
Expert class at an affordable price.
Monocrystal sensors, full-screen display mode, elastography, 3D/4D in a laptop case. Flexible transformation into a stationary scanner with a cart.