Clinical observation of severe methemoglobinemia in a premature newborn
- Model Galina Yurievna
- Tokovaya Inna Anatolyevna
- Eremina Oksana Vasilievna
- Savv Anna Pavlovna
- Shabanova Natalya Evgenevna
- Boykov Sergey Alekseevich
Summary
The article presents a clinical observation of a premature baby with secondary methemoglobinemia, the diagnosis of which was confirmed by a specific laboratory test of blood samples, and also provides a review of current literature data on the pathogenesis, classification and characteristics of clinical manifestations, diagnosis and possibilities of drug treatment of methemoglobinemia.
Key words: methemoglobinemia of newborns, acquired methemoglobinemia
For citation
: Model G.Yu., Tokovaya I.A., Eremina O.V., Savv A.P., Shabanova N.E., Boykov S.A. Clinical observation of severe methemoglobinemia in a premature newborn // Neonatology: news, opinions, training. 2021. T. 7. No. 2. P. 52-58. doi: 10.24411/2308-2402-2019-12004.
Methemoglobinemia (ICD-10: D74) is a heterogeneous group of diseases caused by various etiological and pathogenetic factors resulting from insufficiency of reducing systems in erythrocytes, namely methemoglobin reductases, in which the content of methemoglobin in the blood exceeds the physiological norm (> 1-2% of the total amount hemoglobin) [1].
Classification
I. Primary (hereditary, congenital) methemoglobinemia.
1. Enzymopathic - caused by a sharp decrease or complete absence of the activity of the NADP+-dependent methemoglobin reductase enzyme in erythrocytes.
2. M - hemoglobinopathies (hemoglobinosis M), caused by the presence of unstable or abnormal hemoglobins.
II. Secondary (acquired) methemoglobinemia.
1. Exogenous origin.
2. Endogenous origin [1, 2].
According to the literature, two different reactions occur simultaneously in red blood cells, which balance each other. In one case, the iron in hemoglobin is oxidized, the iron is converted from divalent to trivalent, and methemoglobin is formed, which does not transport oxygen. In the other, methemoglobin is restored back to functionally active hemoglobin. Thus, in healthy people, methemoglobin is in the range of 1-1.5%. The reduction of methemoglobin into active hemoglobin occurs with the help of the red blood cell enzyme NADH-cytochrome b5 reductase and varies from 67 to 73% [1, 3, 4].
Acquired endogenous methemoglobinemia develops with disorders associated with the production and absorption of nitrates during enterocolitis (so-called enterogenous cyanosis). The exact mechanism of development of this form is unknown, but it may be associated with increased endogenous production of nitrites [3].
Acquired exogenous methemoglobinemia occurs when exposed to chemical agents: nitroethane (nail polish remover), aniline (some disinfectants, markers, furacillin), naphthalene, nitric oxide, nitrites (ferryl, amyl, K, Na, isobutyl), nitrates ( converted by bacteria into nitrites); when ingesting certain medications (both in recommended and in increased doses): nitrobenzene derivative (acetaminophen), analgesics (acetanilide, phenacetin), nitrobenzenes/nitrobenzoates, nitroglycerin, nitrofuragin, trinitrotoluene, hydroxylamine, dimethylamine, local anesthetics (lidocaine, prilocaine, benzocaine), dapsone, flutamide, metoclopramide (Cerucal), sulfamethoxazole, sulfonamides, menadione (vitamin K3), naphthoquinone, phenazopuridine (Puridium), antibiotics (ampicillin, amikacin, gentamicin, carbenicillin) [1].
In 1986, E. Jaffe proposed a biochemical classification of congenital enzymopenic methemoglobinemia, according to which 4 types of the disease were identified depending on the nature of the disturbance in the activity of the enzyme NADH-cytochrome b5-reductase in tissues: Type I - benign enzymopenic methemoglobinemia, associated with a deficiency of the cytoplasmic fraction of the enzyme only in red blood cells; Type II - lethal, in addition to methemoglobinemia, is clinically manifested by progressive neurological deficit and is a consequence of a generalized deficiency of the cytoplasmic and membrane-bound forms of the enzyme in all tissues; Type III - clinically similar to type I, caused by impaired activity of the cytoplasmic form of the enzyme in all hematopoietic cells; Type IV is benign and is caused by a deficiency of the enzyme cofactor [2].
The study of methemoglobinemia did not end there, and in 1993 T. Nagai published data that made it possible to equate type III methemoglobinemia with type I [5, 6]. Thus, it was concluded that all types of congenital enzyme methemoglobinemia clinically occur in only 2 variants: benign and progressive lethal. The clinical classification of congenital enzymopenic methemoglobinemia began to look like this:
■ congenital enzymopenic methemoglobinemia type I - benign;
■ congenital enzymopenic methemoglobinemia type II—lethal.
Methemoglobinemia type I benign form was first described in 1845 by the French physician J. Francois [7]. When examining a patient with persistent congenital cyanosis, heart and lung diseases were not identified. And only in 1932 did the first documentary publication on this form of the disease appear [8]. The clinical picture is characterized by cyanosis of the skin and mucous membranes, which is associated with a deficiency of the enzyme NADH-cytochrome b5 reductase located in erythrocytes. After the birth of a child, signs of the disease immediately appear and persist throughout life. In patients, the clinical picture is manifested by periodic headaches, dizziness, shortness of breath, tachycardia, fatigue, drowsiness, and possible retardation in physical and mental development. However, the main concern is cyanosis of the skin as a cosmetic defect. E. Jaffe and D. HuLtquist in 1995 determined that “patients are more blue than sick.” According to laboratory tests, an increase in the content of methemoglobin (15-40%) and the number of red blood cells (compensatory erythrocytosis) is detected.
Thus, an analysis of the literature data shows that methemoglobinemia is a poorly understood disease and the diagnosis is made quite rarely, especially in newborns. The clinical observation of secondary methemoglobinemia in a premature newborn presented below is the first case of an established diagnosis in the Krasnodar region.
Clinical observation
Main diagnosis:
prematurity 30 weeks Postconceptual age 39 weeks. Congenital pneumonia, severe course, convalescent.
Complications:
bronchopulmonary dysplasia, new form, severe course. History of multiple organ dysfunctions. History of persistent pulmonary hypertension. Hyperglycemia while taking contrainsular drugs (history). Necrotizing enterocolitis stage II according to BeLL, convalescent. Hypoxic-hemorrhagic damage to the central nervous system (CNS) in the form of peri-intraventricular hemorrhages of the second degree, early recovery period, depression syndrome. Candidiasis, skin manifestations (history). Anemia of prematurity of mixed origin. Retinopathy of prematurity. Secondary methemoglobinemia.
Concomitant diagnosis:
functioning oval window. Intrauterine malnutrition of the 1st degree (BMI 12%).
Anamnesis
A premature boy from the first pregnancy of a 20-year-old mother, which occurred against the background of the threat of spontaneous miscarriage at the 20th week of pregnancy, requiring hospital treatment. At the gestational age of 24-25 weeks of pregnancy, the threat of premature birth was diagnosed against the background of isthmic-cervical insufficiency, polyhydramnios, and an obstetric pessary was installed. First birth, operative (caesarean section with the use of anesthesia using epidural anesthesia according to the generally accepted method with local anesthetic ropivacaine 0.75%), at a gestation period of 30 weeks. The amniotic fluid is light. The child’s body weight at birth is 1140 g, length is 34 cm, head circumference is 26 cm, chest circumference is 25 cm. Apgar score: 3-4 points. Body weight at transfer - 1180 g (+40 g body weight at birth).
Development of the disease while in the maternity hospital: primary and resuscitation care for the newborn was carried out in the delivery room - radiant heat, prolonged inhalation, artificial ventilation (ALV) through a face mask for 30 s (Peep +5, 21% O2), tracheal intubation, Poractant alfa was administered at a dose of 200 mg/kg, transfer to traditional mechanical ventilation.
The child was admitted to the department in serious condition, in a transport incubator on a traditional ventilator. The severity of the condition was due to respiratory disorders, oxygen dependence, neurological symptoms in the form of central nervous system depression syndrome, morphofunctional immaturity due to prematurity.
In the maternity hospital, the following therapy was carried out: respiratory, antibacterial - ampicillin/sulbactam 100 mg/kg per day, amikacin 15 mg/kg per day; enteral nutrition through an orogastric tube and infusion therapy with the subsidy of partial parenteral nutrition and electrolytes according to physiological need.
On the 5th day of life, the child’s condition remained without any clear dynamics. The premature newborn was transferred to the neonatal intensive care unit No. 2 of the separate department of the Perinatal City of Krasnodar to expand the scope of diagnostic search, treatment and nursing. Main diagnosis: prematurity 30 weeks. Congenital pneumonia against the background of respiratory distress syndrome of the newborn.
Concomitant diagnosis: cerebral ischemia stage II, acute period in the form of central nervous system depression syndrome. Intrauterine malnutrition of the 1st degree (body weight deficiency 12%). Functioning oval window. Patent ductus arteriosus.
Upon admission to the department, the child’s general condition remained severe due to respiratory failure, oxygen dependence requiring respiratory support, intoxication syndrome, neurological symptoms in the form of central nervous system depression syndrome, prematurity, and degree I intrauterine malnutrition. The child was on traditional mechanical ventilation (Puritan Bennett device with parameters: A/C mode; 35 cycles per minute; Pip +18; Peep +5; Tins - 0.32; FiO2 - 40%).
From the moment of admission to the department, the child’s condition attracted attention to pale pink skin with the presence of a “gray color” with a slight cyanotic tint, a venous pattern on the skin of the chest, acrocyanosis, perioral cyanosis, which was regarded as a manifestation of intoxication syndrome. The neurological status included muscle hypotonia, physical inactivity, and depression syndrome. No seizures were recorded. Auscultation in the lungs, against the background of mechanical breathing, weakened breathing was heard, carried out in all parts, and multiple crepitating rales at all points of auscultation. Hemodynamic parameters remained stable. Auscultation at the apex of the heart there is a systolic murmur. The liver is +1.5 cm below the edge of the right costal arch, the spleen was not palpable.
In order to clarify the diagnosis, a complex of laboratory and instrumental studies was carried out.
1. According to chest x-ray, an increase in the pulmonary pattern in the hilar regions of the lower lobes on the right and left and infiltration were determined; there was no differentiation of the caudal part of the roots. There was a diffuse enhancement of the pulmonary pattern due to the interstitial component. The roots of the lungs were poorly structured, with unclear contours. The diaphragm is positioned normally. The diaphragm dome is clear. The heart is not enlarged. The aorta is without features. The mediastinum is not widened. The ribs are not changed, the intercostal spaces are the same on both sides. Conclusion: bilateral pneumonia.
2. Echocardiography: functioning oval window.
3. Neurosonography: peri-intraventricular hemorrhages of the second degree. Forming subependymal cyst on the right. Diffuse increase in echogenicity of the periventricular zones. Asymmetrical dilatation of the lateral ventricles. The resistance index of the anterior cerebral artery during Doppler ultrasound is 0.73.
4. Ultrasound examination of the kidneys: diffuse increase in the parenchyma of both kidneys. Expansion of the collecting system of the right kidney. Echo signs of pyelectasis on the left. The resistance index of the renal arteries during Doppler ultrasound was 0.72.
5. Examination by a neurologist. Diagnosis: hypoxic-hemorrhagic damage to the central nervous system, acute period in the form of peri-intraventricular hemorrhage of the second degree, depression syndrome.
6. Blood type and Rh of the child: B (III) Rh (+) positive. Mother's blood type and Rh: O (I) Rh (-) negative.
7. According to the acid-base composition (ALC): compensated metabolic acidosis, glucose - 4.4 mmol/l, lactate - 2.8 mmol/l, methemoglobin - 1.9%.
8. Complete blood count without pro-inflammatory changes: leukocyte level 16.7x109/l, band neutrophils 9%, segmented neutrophils 33%, hemoglobin - 166 g/l, erythrocytes - 4.7x1012/l, platelets -261x109/l. The capillary glucose level is 3.8 mmol/l.
9. According to the results of a biochemical blood test, the level of total bilirubin was 81.4 µmol/l, the direct fraction was 7.9 µmol/l, C-reactive protein was 0.73 mg/l, transaminases were normal, no electrolyte disturbances were noted.
10. Results of microbiological examination of blood, trachea and anus without growth.
In the department: antibacterial therapy was continued with ampicillin + sulbactam 75 mg/kg per day, amikacin 15 mg/kg per day; respiratory therapy under blood gas monitoring; enteral nutrition and partial parenteral nutrition with subsidies of protein, fat, electrolytes according to physiological needs; protective regime, nursing in incubator conditions, servo control.
On the 8th day of life, the child’s condition progressively worsened in the clinical picture due to the addition of multiple organ dysfunctions in the form of an increase in respiratory disorders and oxygen dependence against the background of progression of hypoxia (Sat 86-84%), requiring transfer to high-frequency oscillatory ventilation (HFOV) with 100 % oxygen (according to acid base balance, decompensated metabolic acidosis, critical hypoxemia, hypercapnia, methemoglobin level was 50%), addition of hemodynamic disorders that required selection of doses of cardiotonic support with stabilization of hemodynamics 4% dopamine at a dose of maximum 10 mcg/kg per minute, adrenaline 1% 1 .0 mcg/kg per minute, clinical picture of necrotizing enterocolitis stage II according to Bell, severe depression of the central nervous system in the neurological status, muscle hypotension, severe anemia (hemoglobin 111 g/l). A blood transfusion with red blood cells depleted of leukocytes and platelets (EMOLT) 0 (I) Rh negative was performed. Against this background, the color of the skin with a progressively increasing pronounced “gray color”, the appearance of central cyanosis, acrocyanosis, cyanosis of the ears as a result of the development of severe hemic hypoxia. In the CBC: leukocytes - 14.7x109 /l, hemoglobin - 111 g/l, hematocrit - 35, platelets 274x109 /l, metamyelocytes - 1%, band neutrophils - 4%, segmented neutrophils - 46%, lymphocytes - 32%, monocytes - 13%, eosinophils - 2%, basophils - 2%.
Taking into account the severity of the condition in the form of multiple organ dysfunctions, the diagnostic search has been expanded to exclude the occurrence of intrauterine infection in the form of late neonatal sepsis. An additional examination of the child and mother was carried out using the method of paired sera to identify the TORCH complex: blood testing using enzyme immunoassay (Tables 1, 2).
The mother and child were found to have positive class G immunoglobulins for toxoplasma infection and cytomegalovirus. When analyzing the results obtained, the titer of maternal antibodies is much higher than that of the child, which indicates transplacental transfer of immunoglobulins from mother to fetus (PCR diagnostics were not performed).
As a passive immunization, normal human immunoglobulin (IgG+IgN+IgA) 5.0 ml/kg per day was added to the treatment.
At the preanalytical stage, a characteristic change in the color of the blood was noted, which acquired a chocolate brown color.
Taking into account literature data [1], an increase in the methemoglobin fraction may indirectly be one of the predictors of the septic process in the body. The child underwent dynamic monitoring of leukocyte counts in a general blood test.
According to the results of the study of a general blood test, attention was drawn to the lack of increase in the level of leukocytes in dynamics and the absence of a shift in the leukocyte formula to the left, while according to the analysis of oxygen status, the level of methemoglobin progressively increased, including at the time of deterioration of the condition (Fig. 1 ).
The CRP level was determined over time. It remained within normal limits, which corresponds to the course of this disease (Table 3).
With spontaneous manifestation, a progressive increase in methemoglobin (2-9-18%) on the 7th day of life reached a critical state (50%) on the 8th day, against the background of normal lactate levels according to the acid-base composition of the blood, with the subsequent development of multiorgan dysfunctions in the form of progression of respiratory failure and oxygen dependence, requiring transfer to high-frequency ventilation, increase in hemodynamic disorders, neurological symptoms in the form of central nervous system depression syndrome against the background of severe central cyanosis. A consultation was held via telemedicine with a hematologist from the Children's Regional Clinical Hospital of the Ministry of Health of the Krasnodar Territory (Krasnodar). The child was diagnosed with idiopathic methemoglobinemia.
As part of the diagnostic search, the presence of markers of a systemic inflammatory response in the child is completely excluded, which is confirmed by the following indicators: negative results when analyzing the level of procalcitonin, the absence of growth of pathological microflora from all loci of the body, a normal level of CRP (Table 3) and the absence of pro-inflammatory changes in the general analysis blood (Fig. 1). Testing for TORCH infections (Tables 1, 2) also gave a negative result. According to the results of the analysis of laboratory data, the increase in methemoglobin levels did not correlate with dynamic lactate indicators, which, in turn, made it possible to exclude neonatal sepsis (Fig. 2). When collecting a family history, there was no evidence of manifestations of methemoglobinemia in the parents and immediate relatives of the child.
5% ascorbic acid was added at a dose of 500 mg/kg per day. In dynamics, against the background of intensive therapy with a step-by-step reduction in the dose of 5% ascorbic acid to 50 mg/kg per day, the level of methemoglobin normalized to 0.8% by the 18th day of life (see Fig. 1, 2), which was the reason for repeated consultation with a hematologist at the Children's Regional Clinical Hospital of the Ministry of Health of the Krasnodar Territory to resolve the issue of canceling pathogenetic therapy. When the level of methemoglobin in the blood increases to 15-20%, it is recommended to add a 1% solution of methylene blue at a dose of 1 mg/kg per day intravenously to therapy. To clarify the form of this disease, the child was consulted at the Federal State Budgetary Institution National Medical Research Center for Pediatric Hematology, Oncology and Immunology named after. Dmitry Rogachev" of the Russian Ministry of Health (Moscow). As a result of the telemedicine consultation, based on the provided medical documentation, the patient was recommended to determine the activity of methemoglobin reductase in erythrocytes. For this purpose, samples of the child’s blood in a volume of 5 ml on an anticoagulant in non-frozen form at a temperature of +4...+6 °C were sent to the laboratory at the Federal State Budgetary Institution National Medical Research Center for Pediatric Hematology, Oncology and Immunology named after. Dmitry Rogachev" of the Russian Ministry of Health. According to a study carried out in the above laboratory, the child was diagnosed with methemoglobinemia as a condition associated with an increased content of methemoglobin - 9.3%. A decrease in the activity of NAD-dependent cytochrome b5 reductase was established - 1.6 units. Act. together with a moderate decrease in Betke coefficients 1.2 (Table 4).
When performing electrophoresis of hemoglobin fractions, the patient revealed abnormal minor fractions of hemoglobin in the zone of γ-chains (zone Z 11 and E). Based on a specific laboratory examination, a conclusion was given: congenital enzymopenic methemoglobinemia was not confirmed, due to the fact that this finding may be a consequence of the child’s prematurity.
Most likely, in this patient, methemoglobinemia was of a secondary (acquired) nature, which is confirmed by the results of differential diagnosis of hereditary methemoglobinemia (see Table 4). In order to exclude a drug-induced cause of the development of secondary methemoglobinemia, a detailed analysis of drug therapy was carried out. No drugs containing nitro- and amino groups used in therapy for this child have been identified.
During his stay in the department, the child's condition stabilized. On the 30th day of life, he was transferred to non-invasive ventilation for 8 days, followed by transfer to spontaneous breathing with additional supply of humidified oxygen through binasal cannulas with a FiO2 concentration of 30%. At 2 months of life, in a stable condition, he was transferred from the intensive care unit No. 2 to the pediatric department of the Children's Regional Clinical Hospital of the Ministry of Health of the Krasnodar Territory for the purpose of further treatment and nursing.
On the 74th day of life, the child was discharged home in satisfactory condition under the supervision of the follow-up department of the Children's Consultative and Diagnostic Center in Krasnodar.
Discussion
Methemoglobinemia is a rare disease (according to some data, to date, only about 600 cases of methemoglobinemia have been described in the world), the clinical picture of which is nonspecific and can occur in combination with other symptoms, which makes diagnosis difficult. Based on the combination of a number of symptoms in combination with laboratory examination methods, this clinical case represents an urgent problem, taking into account the small amount of information on this condition in modern neonatology. Neonatologists should be alert to this pathology and have the opportunity to timely diagnose and refer for consultation to related specialists in order to clarify the form of this disease, which may provide a favorable prognosis.
Conflict of interest
. The authors declare no conflict of interest.
Literature
1. Kazanets E.G. Methemoglobinemia // Children's Hospital. 2009. No. 1. P. 38-42.
2. Jaffe ER Enzymopenic hereditary methemoglobinemia: a clinical/biochemical classification // Blood Cells. 1986. Vol. 12, N 1. P. 81-90.
3. Kazanets E.G., Andreeva A.P., Khangulov S.V., Tokarev Yu.N. Hereditary cyanosis caused by the presence of abnormal hemoglobins of group M in the blood: detection, identification, properties // Hematol. and transfusiol. 1990. No. 3. P. 9-13.
4. Kleimenova I.S., Shvyrev A.P., Serednyak V.G. Sotnikova N.A. and others. Congenital enzymopenic methemoglobinemia type II // Ros. Vestn. perinatol and pediatrician. 2011. No. 6. P. 80-87.
5. Nagai T., Shirabe K., Yubisui T., Takeshita M. Analysis of mutant NADH-cytochrome-b5-reductase: apparent “type III” methemoglobinemia can be explained as type I with an unstable reductase // Blood. 1993. Vol. 81. P. 808-814.
6. Tanishima K., Tanimoto K., Tomoda A. et al. Hereditary methemoglobinemia due to cytochrome-b5-reductase deficiency in blood cells without associated neurologic and mental disorders // Blood. 1985. Vol. 66. P. 1288-1291.
7. Francois J. Cas de cyanose congenitale sans cause apparente // Bull. Acad. Roy Med. Belg. 1845. Vol. 4. P. 698.
8. Hitzenberger K. Autotoxische zyanose (intraglobulare methamoglo-binamie) // Wien. Arch. Intern. Med. 1932. Vol. 23. P. 85-96.
References
1. Kazanets EG Methemoglobinemia. Detskaya bol'nitsa [Children's Hospital]. 2009; (1): 38-42 (in Russian)
2. Jaffe ER Enzymopenic hereditary methemoglobinemia: a clinical/biochemical classification. Blood Cells 1986; 12 (1): 81-90.
3. Kazanets EG, Andreeva AP Khagulov SV, Tokarev Yu.N. Hereditary cyanosis caused by anomaly hemoglobin group M in blood: revealing, identification, properties. Gematologiya i transfuziologiya. 1990; (3): 9-13. (in Russian)
4. Kleimenova IS, Shvirev AP Serednyak VG, Sotnikova NA, et al. Congenital enzymopenic methemoglobinemia of type II. Rossiyskiy vestnik perinatologii i pediatrii. 2011; (6): 80-7. (in Russian)
5. Nagai T., Shirabe K., Yubisui T., Takeshita M. Analysis of mutant NADH-cytochrome-b5-reductase: apparent “type III” methemoglobinemia can be explained as type I with an unstable reductase. Blood. 1993; 81:808-14.
6. Tanishima K., Tanimoto K., Tomoda A., et al. Hereditary methemoglobinemia due to cytochrome-b5-reductase deficiency in blood cells without associated neurological and mental disorders. Blood. 1985; 66: 1288-91.
7. Francois J. Cas de cyanose congenitale sans cause apparente. Bull Acad Roy Med Belg. 1845; 4: 698.
8. Hitzenberger K. Autotoxische zyanose (intraglobulare methamoglo-binamie). Wien Arch Intern Med. 1932; 23: 85-96.
Hereditary enzymopenic
methemoglobinemia is a hereditary disease in which the content of methemoglobin (MetHb) in the blood exceeds the physiological norm (> 1–2% of the total amount of Hb) (Kazanets E.G., 2009). In Russia, it is observed mainly in adults in the form of endemic foci in Yakutia in the area of the Vilyuy River (Tokarev Yu.N. et al., 1975, 1976, 1983; Derviz G.V., 1977; Nisan L.G. et al., 1987 ). The severity of symptoms is determined by the amount of methemoglobin in the blood. An increase in MetHb to 10% most often does not produce clinically significant manifestations. When MetHb increases within 10–20%, cyanosis of the mucous membranes and skin appears, general weakness, malaise, memory loss, irritability, and headaches occur. When the MetHb content is within 30–50%, the above symptoms are accompanied by pain in the heart of various types, shortness of breath, dizziness, pronounced cyanosis, and increased blood viscosity. MetHb content of more than 70% is incompatible with life.
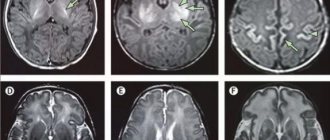
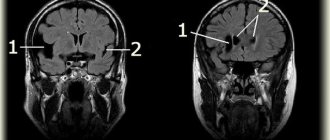
| Symptoms: a) moderate cyanosis of the lips and oral mucosa, manifestations of dystonia in the facial muscles; b) cyanosis of the nail plates. For methemoglobinemia type 1 patients periodically experience headaches, dizziness, shortness of breath, tachycardia, fatigue, drowsiness, and possibly retardation in physical and mental development. For methemoglobinemia type 2 cyanosis is accompanied by delayed intellectual development, progressive secondary microcephaly, and impaired development of the nervous system. MRI often reveals cortical and subcortical atrophy. Laboratory research methods reveal an increase in the content of methemoglobin (1540%) and the number of red blood cells (compensatory erythrocytosis). |
Molecular genetic cause of the disease
— mutations in the DIAI gene, encoding two forms of the enzyme NADH-cytochrome b5 reductase. The membrane-bound form of the enzyme is involved in the basic biochemical processes of each cell, the soluble form is involved in the reduction of methemoglobin in erythrocytes. When mutations lead to disruption of the functioning of only the soluble form of the enzyme, type 1 of the disease occurs. With mutations that disrupt the functioning of both forms of the enzyme - the second type. In Russia, type 1 methemoglobinemia is most common among the Sakha people; its frequency in Yakutia is 1:5700 people, i.e. every 37 Yakuts are heterozygous carriers of the disease. The molecular genetic cause of the disease in the Yakut variant of methemoglobinemia type 1 is the c.806C›T mutation in the DIA1 gene, leading to the Pro269Leu amino acid substitution.
The role of red blood cells in the metabolism of methemoglobin was established by Gibson QH in 1943-48. According to modern views, two opposite reactions occur simultaneously in red blood cells, balancing each other. On the one hand, the iron of hemoglobin is oxidized, turning from divalent to trivalent, and oxygen- intolerant methemoglobin (MetHb) is formed; 0.5-3% of the total amount of hemoglobin in the body is formed per day (Derviz G.V., 1977) . On the other hand, this MetHb is constantly restored back into functionally active hemoglobin, as a result, in healthy people the MetHb level is kept within the range of 1-1.5%. The process of MetHb reduction in erythrocytes has been studied quite well. In erythrocytes, several reducing systems are known that are far from equivalent in their effectiveness. 67-73% of the restoration of MetHb to active Hb is provided by the erythrocyte enzyme NADH-cytochromeb5 reductase (NAD H2-MetHb reductase, NADH ferricyanide reductase, NADH dehydrogenase, diaphorase I, NADH dehydrogenase, its role was established in 1959 by Scott and Griffith). When this system is blocked due to genetic defects, minor pathways of direct reduction of MetHb are stimulated by endogenous reducing agents (ascorbic acid, reduced glutathione, flavin, tetrahydropterin, cysteine, tryptophan metabolites) or other systems (Kazanets E.G., 2009).
Hereditary enzymopenic methemoglobinemia (HEM), or methemoglobinemia caused by deficiency of NADH-cytochrome-Lb-reduetase (MW 250800) still remains a poorly studied pathology, especially in children (Jaffe E R., 1986; Shirabe K., Yubisui T., 1991; Wu Y., Mota LV, Kaplan JC, 1995; Chang-Hui Huang, 1998; Dekker J., Eppink M., 2001). The pathogenesis of clinical manifestations of NEM is determined by chronic hypoxia due to the oxidation of part of hemoglobin into the meta-form and the formation of “valence” hybrids that are unable to capture oxygen in the lungs and deliver it to tissues (Andreeva A.P., 1976, 1977; Zakharova F.A., 1982 ; Tokarev Yu.N., 1983). The degree of severity of clinical symptoms depends on the content of methemoglobin in the blood and the compensatory abilities of the cardiovascular, respiratory and hematopoietic systems in the process of adaptation to hypoxia (Kushakovsky M.S., 1968; Nissan L.G., 1987; Askerova T.A., 1995 ).
In the literature there are descriptions of the clinical picture of the disease and the condition of peripheral blood in adults (Zakharova F.A., 1982; Tokarev Yu.N., 1983; Jenkins JM, 1992; Shirabe K., 1995; Wang Y., Wu Y., 2000 ). At the same time, an increase in the content of hemoglobin and red blood cells was established, obviously of a compensatory nature (Tokarev Yu. N., 1980, 1983). Zakharova F.A. (1982) reports an increase in serum iron levels in patients. However, there is no data in the literature on the functional and morphological state of the peripheral erythron unit in this disease. There is only isolated information about changes in the morphological characteristics of erythrocytes in various hemoglobinopathies accompanied by hypoxia (Kovalyova L.G., Postnikov Yu.V., 1987; Kazanets E.G., 1990; Troitskaya O.V., 1996, 1999; Nagai T. ., 1980).
The clinical picture of hereditary enzymatic methemoglobinemia in children has age-related characteristics: schoolchildren are characterized by more pronounced cyanosis, symptoms of functional cardiopathy and hypoxia. Indicators of body weight and height in children with hereditary enzymopenic methemoglobinemia are below average, and their deficiency increases with age. The peripheral link of erythrone in patients, regardless of age, is characterized by an increase in the number of immature forms of erythrocytes, as well as a significant increase in degenerative and flat forms of erythrocytes. Patients with hereditary enzymopenic methemoglobinemia, regardless of age, had increased lipid peroxidation and decreased activity of antioxidant systems - catalase and low molecular weight antioxidants. The younger group of children is characterized by an increase in superoxide dismutase. Pathogenetic therapy with ascorbic acid leads to improvement. condition of children, a decrease in the level of methemoglobin and an increase in the activity of total antioxidants.
Course of the disease
, as a rule, benign. The life expectancy of patients is not affected. The blood of such patients is dark brown as a result of increased methemoglobin content. In some cases, untreated patients may experience secondary compensatory erythrocytosis, an increase in hemoglobin (up to 170-240 g/l), slight reticulocytosis (less than 3%), an increase in blood viscosity and a decrease in ESR. There may be a slight increase in bilirubin in the blood serum due to the indirect (free) fraction. In heterozygotes, the concentration of methemoglobin in the blood is 12%, there are no signs of the disease. Cyanosis may appear after taking methemoglobin-forming drugs (Troshin V.A., 2007; Kazanets E.G., 2009).
In Yakutia, an unusually high prevalence of type I NEM has been registered among the indigenous inhabitants of the republic (Tokarev Yu.N., 1983, where) its frequency is 1:5700 people, i.e. Every 37 Yakuts are heterozygous carriers of the disease. In fact, Yakutia is the only focus of this disease in Russia. (Outside Russia, hereditary benign methemoglobinemia is common among residents of Greenland, Alaska Indians and members of the Navajo tribe in the USA).
It is possible that the accumulation of this disease occurred due to the founder effect - a population genetic phenomenon associated with limited reproductive numbers in the past of fairly isolated populations (Seroshevsky V.L. 1993). A molecular genetic study of Yakut patients with NEM did not confirm the presence of 3 point mutations in the NADH-cytochrome b5 reductase gene (Ag5701n, Leu72Pro, Ya1105Me1), which are found in the Japanese and Chinese in Asia.
Literature
- Blumenfeld L.A. Hemoglobin and reversible addition of oxygen. M.: Sov. science, 1957.
- Derviz G.V. Hereditary enzymopenic methemoglobinemia.//Clinical Medicine, 1977, No. 5, p. 8.
- Kazanets E.G. Methemoglobinemia.// Children's Hospital, 2009, No. 1, pp. 38-42.
- Kazanets E.G., Andreeva A.P., Khangulov S.V., Tokarev Yu.N. Hereditary cyanosis caused by the presence of abnormal hemoglobins of group M in the blood: detection, identification, properties // Hematology and Transfusiology, 1990, No. 3, p. 9–13.
- Krasnopolskaya K.D. Hereditary metabolic diseases. – M., 2005, 290 p.
- Melnik A.I., Melnik V.A. Exacerbation of hereditary methemoglobinemia in infant twins // Pediatrics. - 1986. - No. 12. - P. 58 - 60.
- Nisan L.G., Gurevich S.P., Kazanets E.G., Frolova M.I., Tokarev Yu.N., Salmova T.S., Butina M.V. Hereditary enzymopenic methemoglobinemia in newborns // Questions of motherhood and childhood, 1987, No. 1, p. 74–75.
- Tokarev Yu.N., Fainshtein F.E. and others//Problems of hematology and transfusiology. T.2. M., 1976, pp. 123-128.
- Tokarev Yu.N., Hollan S.R., Corral-Almonte H.S.// Hereditary anemia and hemoglobinopathies. – M., 1983.
- Torshin V. A. Clinically significant dysmoglobins. Carboxyhemoglobin // Laboratory. 2007. No. 1. P. 17-18.
- Chelnokov S.B., Yakovleva E.A., Pudina N.A. A case of severe methemoglobinemia in a premature newborn baby // Bulletin of Intensive Care. 2002. No. 2. S.1821.
- Aalfs CM, Salieb-Beugelaar GB, Wanders RJ, Mannens MM, Wijburg FA. A case of methemoglobinemia type II due to NADH-cytochrome b5 reductase deficiency: determination of the molecular basis. Hum Mutat 2000;16:18–22.
- Abdemula MS. La methemoglobinemia hereditaire recessive de type II. A propos d'une observation. Revue Maghrebine de Pediatrie 2002; XII-IV:207–10.
- Bewley MC, Davis CA, Marohnic CC, Taormina D, Barber MJ. The structure of the S127P mutant of cytochrome b5 reductase that causes methemoglobinemia shows the AMP moiety of the flavin occupying the substrate binding site. Biochemistry 2003; 42:13145–51.
- Blumenfeld LA Physics of Bioenergetic Processes. NY: Springer-Verlag, 1983.
- Davis CA, Barber MJ. Cytochrome b5 oxidoreductase: expression and characterization of the original familial idiopathic methemoglobinemia mutations E255 and G291D. Arch Biochem Biophys 2004; 425:123–32.
- Dekker J, Eppink MH, van Zwieten R, de Rijk T, Remacha AF, Law LK, et al. Seven new mutations in the nicotinamide adenine dinucleotide reduced-cytochrome b(5) reductase gene leading to methemoglobinemia type I. Blood 2001;97:1106–14.
- Den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000;15:7–12.
- Dickerson RE, Geis I. Hemoglobin: Structure, Function, Evolution, and Pathology (Benjamin Cummings: Menlo Park, CA), 1983.
- Du M, Shirabe K, Takeshita M. Identification of alternative first exons of NADH-cytochrome b5 reductase gene expressed ubiquitously in human cells. Biochem Biophys Res Commun1997; 235:779–83.
- Ewenczyk C., Leroux A., Roubergue A., Laugel V., Afenjar A., Saudubray JM, Beauvais P., BillettedeVillemeur T., Vidailhet M., Roze E. Recessive hereditary metaemoglobinaemia, typeII: delineation of the clinical spectrum. / Brain (2008), 131,760-771.
- Fermo E, Bianchi P, Vercellati C, et al. Recessive hereditary methemoglobinemia: Two novel mutations in the NADH-cytochrome b5 reductase gene. /J Blood Cells Mol Dis 2008 Mar 15.
- Fisher RA, Povey S, Bobrow M, Solomon E, Boyd Y, Carritt B. Assignment of the DIA1locus to chromosome 22. Ann Hum Genet 1977;41:151–5.
- Francois. Cas de cyanose conge?nitale sans cause apparente. Bull Acad. Roy Med Belg 1845; 4: 698.
- Fusco C, Soncini G, Frattini D, Giustina ED, Vercellati C, Fermo E, Bianchi P. Cerebellar atrophy in a child with hereditary methemoglobinemia type II./ Brain Dev 2010 Jul 21.
- Gibson QH. The reduction of methaemoglobin in red blood cells and studies on the cause of idiopathic methaemoglobinaemia. Biochem J 1948; 42:13–23.
- Gokalp S, Unuvar E, Oguz F, Kilic A, Sidal M. A case with quadriparetic cerebral palsy and cyanosis: congenital methemoglobinemia. Pediatr Neurol 2005; 33:131–3.
- Gonzalez R, Estrada M, Wade M, de la Torre E, Svarch E, Fernandez O, et al. Heterogeneity of hereditary methaemoglobinaemia: a study of 4 Cuban families with NADH-Methaemoglobin reductase deficiency including a new variant (Santiago de Cuba variant). Scand J Haematol 1978; 20:385–93.
- Grabowska D, Plochocka D, Jablonska-Skwiecinska E, Chelstowska A, Lewandowska I, Staniszewska K, et al. Compound heterozygosity of two missense mutations in the NADH-cytochrome b5 reductase gene of a Polish patient with type I recessive congenital metaemoglobinaemia. Eur J Haematol 2003; 70:404–9.
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18:2714–23.
- Hegesh E, Hegesh J, Kaftory A. Congenital methemoglobinemia with a deficiency of cytochrome b5. N Engl J Med. 1986; 314:757–761.
- Heusden A, Willems C, Lambotte C, Hainaut H, Chapelle P, Malchair R. Arch Fr Pediatr 1971; 28: 631–45.
- Higasa K, Manabe JI, Yubisui T, Sumimoto H, Pung-Amritt P, Tanphaichitr VS, et al. Molecular basis of hereditary metaemoglobinaemia, types I and II: two novel mutations in the NADH-cytochrome b5 reductase gene. Br J Haematol 1998; 103:922–30.
- Hildebrandt A, Estabrook RW. Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Arch Biochem Biophys 1971; 143:66–79.
- Hirono H. Lipids of liver, kidney, spleen and muscle in a case of generalized deficiency of cytochrome b5 reductase in congenital methemoglobinemia with mental retardation. Lipids 1984; 19:60–3.
- Hitzenberger K. Autotoxische zyanose (intraglobulare methamoglobinamie). Wien Arch Intern Med.1932;23:85.
- Hultquist DE, Passon PG. Catalysis of methaemoglobin reduction by erythrocyte cytochrome b5 and cytochrome b5 reductase. Nature1971; 229:252–4.
- Jablonska-Skwiecinska E, Holtorp-Tyszkiewiczowa J, Staniszewska K. Generalized deficiency of the NADH-methemoglobin reductase in congenital methemoglobinemia with neurological symptoms. Biomed Biochim Acta 1984; 43:S98–100.
- Jaffe ER, Hultquist DE. Cytochrome b5 reductase deficiency and Enzymopenic hereditary methemoglobinemia. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Basis of Inherited Disease.7th ed. NewYork, NY: McGraw-Hill;1995:2267–2280.
- Junien C, Leroux A, Lostanlen D, Reghis A, Boue J, Nicolas H, et al. Prenatal diagnosis of congenital Enzymopenic metaemoglobinaemia with mental retardation due to generalized cytochrome b5 reductase deficiency: first port of two cases. Prenat Diagn 1981; 1:17–24.
- Kaftory A, Freundlich E, Manaster J, Shukri A, Hegesh E. Prenatal Diagnosis of congenital methemoglobinemia with mental retardation. Isr J Med Sci 1986; 22:837–40.
- Kobayashi Y, Fukumaki Y, Yubisui T, Inoue J, Sakaki Y. Serine-proline replacement at residue 127 of NADH-cytochrome b5 reductase causes hereditary methemoglobinemia, generalized type. Blood 1990; 75:1408–13.
- Kugler W, Pekrun A, Laspe P, Erdlenbruch B, Lakomek M. Molecular basis of recessive congenital methemoglobinemia, types I and II: Exon skipping and three novel missense mutations in the NADH-cytochrome b5 reductase (diaphorase 1) gene. Hum Mutat 2001; 17:348.
- Lamy M, Frezal J, Jammet ML, Josso N. Nouv Rev Fr Hematol 1963; 3: 105–20.
- Leroux A, Mota Vieira L, Kahn A. Transcriptional and translational mechanisms of cytochrome b5 reductase isoenzyme generation in humans. Biochem J 2001; 355:529–35.
- Lostanlen D, Vieira de Barros A, Leroux A, Kaplan JC. Soluble NADH-cytochrome b5 reductase from rabbit liver cytosol: partial purification and characterization. Biochim Biophys Acta 1978; 526:42–51.
- Manabe J, Arya R, Sumimoto H, Yubisui T, Bellingham AJ, Layton DM, et al. Two novel mutations in the reduced nicotinamide adenine dinucleotide (NADH)-cytochrome b5 reductase gene of a patient with generalized type, hereditary methemoglobinemia. Blood 1996; 88: 3 208 – 15.
- Maran J, Guan Y, Ou CN, Prchal JT. Heterogeneity of the molecular biology of methemoglobinemia: a study of eight consecutive patients. Haematologica 2005; 90:687–9.
- Nussenzveig RH, Lingam HB, Gaikwad A, Zhu Q, Jing N, Prchal JT. A novel mutation of the cytochrome-b5 reductas gene in an Indian patient: the molecular basis of type I methemoglobinemia. Haematologica 2006; 91:1542–5.
- Orsini A, Vovan L, Brusquet Y, Gabriel B, Sebag F, Galtier M. Congenital methemoglobinemia due to NADH (DPNH) dependent methemoglobin reductase deficiency. Mars Med 1972; 109:279–81.
- Owen EP, Berens J, Marinaki AM, Ipp H, Harley EH. Recessive congenital metaemoglobinaemia type II a new mutation which causes incorrect splicing in the NADH-cytochrome b5 reductase gene. J Inherit Metab Dis 1997; 20: 610.
- Percy MJ, Crowley LJ, Davis CA, McMullin MF, Savage G, Hughes J, et al. Recessive congenital metaemoglobinaemia: functional characterization of the novel D239G mutation in the NADH-binding lobe of cytochrome b5 reductase. Br J Haematol 2005; 129:847–53.
- Ronconi G, Ferracin G. Riv Clin Pediatr 1964; 74: 152–9.
- Roussel A, Maestraggi P, Tremoulet M, Marchand. A new case of recessive congenital methemoglobinemia. Arch Fr Pediatr 1963; 20: 745–50.
- Sacerdotti-Favini. Methemoglobinemia costituzionale con cerebropatia e oligofrenia. Acta pediat Lat 1948; 11:255.
- Scott EM, Griffith IV. The enzyme defect of hereditary methemoglobinemia: diaphorase. Biochim Biophys Acta.1959;34:584–586.
- Shirabe K, Landi MT, Takeshita M, Uziel G, Fedrizzi E, Borgese N. A novel point mutation in a 3'splice site of the NADH-cytochrome b5 reductase gene results in immunologically undetectable enzyme and impaired NADH-dependent ascorbate regeneration in cultured fibro-blasts of a patient with type II hereditary methemoglobinemia. Am J Hum Genet 1995; 57:302–10.
- Shonola S. Da-Silva, Imran S. Sajan and Joseph P. Underwood, III. Congenital Methemoglobinemia: A Rare Cause of Cyanosis in the Newborn Case Report./ Pediatrics, 2003;112;e158-e161.
- Shotelersuk V, Tosukhowong P, Chotivitayatarakorn P, Pongpunlert W. A Thai boy with hereditary Enzymopenic methemoglobinemia type II. J Med Assoc Thai 2000; 83:1380–6.
- Smith, R.P. Chapter 11, Toxic responses of the blood. In Casarett and Doull's Toxicology. The Basic Science of Poisons, 5th ed., CD Klaassen, Ed., McGraw-Hill, New York, 1996.
- Strittmatter P, Spatz L, Corcoran D, Rogers MJ, Setlow B, Redline R. Purification and properties of rat liver microsomal stearyl coenzyme A desaturase. Proc Natl Acad Sci USA 1974; 71:4565–9.
- Takeshita M, Matsuki T, Tanishima K, Yubisui T, Yoneyama Y, Kurata K, et al. Alteration of NADH-diaphorase and cytochrome b5 reductase activities of erythrocytes, platelets, and leukocytes in hereditary metaemoglobinaemia with and without mental retardation. J Med Genet 1982; 19: 204–9.
- Toelle SP, Boltshauser E, Mossner E, Zurbriggen K, Eber S. Sever neurological impairment in hereditary metaemoglobinaemia type 2. Eur J Pediatr 2004; 163:207–9.
- Trost C. The blue people of Troublesome Creek. Science 82, November, pp. 35-39, 1982.
- Vieira M, Kaplan JC, Kahn A, Leroux A. Four new mutations in the NADH-cytochrome b5 reductase gene from patients with recessive congenital methemoglobinemia type II. Blood 1995; 85:2254–62.
- Wang Y, Wu YS, Zheng PZ, Yang WX, Fang GA, Tang YC, et al. A novel mutation in the NADH-cytochrome b5 reductase gene of a Chinese patient with recessive congenital methemoglobinemia. Blood 2000; 95:3250–5.
- Williamson DA, Black JA. Congenital metaemoglobinaemia; a case report. Great Ormond St J 1954; 7:56–61.
- Wu YS, Wang Y, Huang CH, Lan FH, Zhu ZY. A compound heterozygote in the NADH-cytochrome b5 reductase gene from a Chinese patient with hereditary methemoglobinemia type I. Int J Hematol 2000;72: 34–6.
- Yawata Y, Ding L, Tanishima K, Tomoda A. New variant of cytochrome b5 reductase deficiency (b5RKurashiki) in red cells, platelets, lymphocytes, and cultured fibroblasts with congenital methemoglobinemia, mental and neurological retardation, and skeletal anomalies. Am J Hematol 1992; 40: 299–305.
- Yilmaz D, Cogulu O, Ozkinay F, Kavakli K, Roos D. A novel mutation in the DIA1 gene in a patient with methemoglobinemia type II. Am J Med Genet A 2005; 133:101–2.
- Yuksel D, Senbil N, Yilmaz D, Yarali N, Gurer YK A rare cause of mental motor retardation: recessive congenital methemoglobinemia type II. /Turk J Pediatr 2009 Mar-Apr; 51(2):187-9.
- Zorc J, Kanic Z. A cyanotic infant: true blue or otherwise? Pediatr Ann 2001;30:597–601.
Methemoglobinemia
Methemoglobinemia is a condition characterized by an increased content of methemoglobin (oxidized hemoglobin) in the blood and tissue hypoxia. The development of methemoglobinemia is accompanied by acrocyanosis, weakness, headaches, dizziness, palpitations, and shortness of breath on exertion. A characteristic sign of methemoglobinemia is the brown-chocolate color of the blood. To confirm the diagnosis, an assessment of symptoms, laboratory studies and tests are carried out. In case of severe methemoglobinemia, oxygen therapy, administration of ascorbic acid, methylene blue solution, and in some cases, exchange blood transfusion are indicated. Symptoms of methemoglobinemia
Signs of hereditary methemoglobinemia become noticeable during the neonatal period. Cyanosis is noticeable on the skin and visible mucous membranes of the child (in the area of the lips, nasolabial triangle, earlobes, nail bed). In addition to hereditary methemoglobinemia, other congenital anomalies are often detected in children - changes in the configuration of the skull, underdevelopment of the upper limbs, vaginal atresia, thalassemia, etc. Children are often lagging behind in psychomotor development. Depending on the level of the MtHb fraction, the severity of manifestations of congenital and acquired methemoglobinemia can vary significantly.
At the concentration of MtHb in the blood:
- 3-15% - the skin acquires a grayish tint
- 15-30% - cyanosis develops, the blood becomes chocolate brown
- 30-50% - weakness, headache, tachycardia, shortness of breath on exertion, dizziness, fainting occur
- 50-70% - arrhythmia, rapid breathing, convulsions occur; metabolic acidosis develops; there are signs of central nervous system depression, coma is possible
- >70% - severe hypoxia, death.
Any form of methemoglobinemia is characterized by a slate-gray coloration of the skin, but there are no “drum stick” changes in the nail phalanges characteristic of cardiopulmonary diseases.
Acrocyanosis increases with cooling, eating nitrate-containing foods, with toxicosis of pregnancy in women, as well as taking methemoglobin-forming medications. Treatment and prevention of methemoglobinemia
Patients with no clinical manifestations do not require special therapy. If there is a significant concentration of MtHb in the blood and extensive symptoms of methemoglobinemia, drug therapy is prescribed to promote the conversion of methemoglobin into hemoglobin. Ascorbic acid and methylene blue (chromosmon) have such restorative properties. Ascorbic acid is prescribed orally, first in large doses, and as the condition normalizes - in maintenance doses. A solution of methylene blue is administered intravenously. In case of severe cyanosis, oxygen therapy is performed. Severe methemoglobinemia is an indication for exchange transfusion. The course of hereditary and drug-induced methemoglobinemia is usually benign. An unfavorable outcome is possible in severe forms of toxic methemoglobinemia with a high content of MtHb in erythrocytes. Patients with this pathology should avoid contact with methemoglobin-forming substances, hypothermia and other provoking factors. Prevention of congenital methemoglobinemia involves conducting medical genetic consultation to identify heterozygous carriers among future parents.