Клиническое наблюдение тяжелой метгемоглобинемии у недоношенного новорожденного
- Модель Галина Юрьевна
- Токовая Инна Анатольевна
- Еремина Оксана Васильевна
- Савв Анна Павловна
- Шабанова Наталья Евгеньевна
- Бойков Сергей Алексеевич
Резюме
В статье представлено клиническое наблюдение недоношенного ребенка с вторичной метгемоглобинемией, диагноз которой был подтвержден специфическим лабораторным исследованием образцов крови, а также проведен обзор актуальных данных литературы о патогенезе, классификации и особенностях клинических проявлений, диагностике и возможностях медикаментозного лечения метгемоглобинемий.
Ключевые слова: метгемоглобинемия новорожденных, приобретенная метгемоглобинемия
Для цитирования
: Модель Г.Ю., Токовая И.А., Еремина О.В., Савв А.П., Шабанова Н.Е., Бойков С.А. Клиническое наблюдение тяжелой метгемоглобинемии у недоношенного новорожденного // Неонатология: новости, мнения, обучение. 2021. Т. 7. № 2. С. 52-58. doi: 10.24411/2308-2402-2019-12004.
Метгемоглобинемии (МКБ-10: D74) — гетерогенная группа заболеваний, обусловленных различными этиологическими и патогенетическими факторами, возникающими в результате недостаточности восстанавливающих систем в эритроцитах, а именно метгемоглобинредуктаз, при которых содержание метгемоглобина в крови превышает физиологическую норму (>1-2% общего количества гемоглобина) [1].
Классификация
I. Первичные (наследственные, врожденные) метгемоглобинемии.
1. Энзимопатические — обусловленные резким снижением или полным отсутствием в эритроцитах активности фермента НАДФ+-зависимой метгемоглобинредуктазы.
2. М — гемоглобинопатии (гемоглобиноз М), обусловленные наличием нестабильных или аномальных гемоглобинов.
II. Вторичные (приобретенные) метгемоглобинемии.
1. Экзогенного происхождения.
2. Эндогенного происхождения [1, 2].
По данным литературы, в эритроцитах одновременно происходят 2 разные реакции, которые уравновешивают друг друга. В одном случае железо гемоглобина окисляется, происходит превращение железа из двухвалентного в трехвалентное и образуется метгемоглобин, который не переносит кислород. В другом — метгемоглобин восстанавливается обратно в функционально активный гемоглобин. Таким образом, у здоровых людей метгемоглобин находится в пределах до 1-1,5%. Восстановление метгемоглобина в активный гемоглобин происходит с помощью фермента эритроцитов НАДН-цитохром-b5-редуктаза и варьирует от 67 до 73% [1, 3, 4].
Приобретенная метгемоглобинемия эндогенная развивается при нарушениях, связанных с продукцией и всасыванием нитратов при энтероколитах (так называемый энтерогенный цианоз). Точный механизм развития такой формы неизвестен, но, возможно, он связан с повышенной эндогенной продукцией нитритов [3].
Приобретенная метгемоглобинемия экзогенная возникает при воздействии химических агентов: нитроэтан (жидкость для снятия лака с ногтей), анилин (некоторые средства дезинфекции, маркеры, фурациллин), нафталин, окись азота, нитриты (феррил, амил, К, Na, изобутил), нитраты (превращаемые бактериями в нитриты); при приеме внутрь некоторых лекарственных средств (как в рекомендованных, так и в повышенных дозах): производное нитробензола (ацетаминофен), анальгетики (ацетанилид, фенацетин), нитробензолы/нитробензоаты, нитроглицерин, нитрофурагин, тринитротолуол, гидроксиламин, диметиламин, местные анестетики (лидокаин, прилокаин, бензокаин), дапсон, флютамид, метоклопрамид (церукал), сульфаметоксазол, сульфаниламиды, менадион (витамин К3), нафтоквинон, феназопуридин (пуридиум), антибиотики (ампициллин, амикацин, гентамицин, карбенициллин) [1].
В 1986 г. E. Jaffe предложил биохимическую классификацию врожденных энзимопенических метгемоглобинемий, согласно которой выделено 4 типа заболевания в зависимости от характера нарушения активности фермента НАДН-цитохром-Ь5-редуктазы в тканях: I тип — доброкачественная энзимопеническая метгемоглобинемия, связанная с дефицитом цитоплазматической фракции фермента только в эритроцитах; II тип — летальный, помимо метгемоглобинемии, клинически проявляется прогрессирующим неврологическим дефицитом и является следствием генерализованного дефицита цитоплазматической и мембраносвязанной форм фермента во всех тканях; III тип — клинически сходный с I типом, вызван нарушением активности цитоплазматической формы фермента во всех гемопоэтических клетках; IV тип — доброкачественный, вызван дефицитом кофактора фермента [2].
На этом изучение метгемоглобинемии не завершилось, и в 1993 г. T. Nagai опубликовал данные, которые позволили приравнять III тип метгемоглобинемии к I типу [5, 6]. Таким образом, был сделан вывод, что все типы врожденной энзи-мопенической метгемоглобинемии клинически протекают только в 2 вариантах: доброкачественном и прогрессирующем летальном. Клиническая классификация врожденных энзимопенических метгемоглобинемий стала выглядеть следующим образом:
■ врожденная энзимопеническая метгемоглобинемия I типа — доброкачественная;
■ врожденная энзимопеническая метгемоглобинемия II типа — летальная.
Впервые метгемоглобинемия I типа доброкачественной формы была описана в 1845 г. французским врачом J. Francois [7]. При обследовании больного со стойким врожденным цианозом у него не были установлены заболевания сердца и легких. И только в 1932 г. появляется первая документальная публикация по данной форме заболевания [8]. Клиническая картина характеризуется синюшностью кожи и слизистых оболочек, которая связана с дефицитом фермента НАДН-цитохром-Ь5-редуктазы, расположенного в эритроцитах. После рождения ребенка сразу появляются признаки заболевания, которые сохраняются в течение всей жизни. У больных клиническая картина проявляется периодическими головными болями, головокружением, одышкой, тахикардией, быстрой утомляемостью, сонливостью, возможно отставание в физическом и психическом развитии. Однако преимущественно беспокоит цианоз кожных покровов как косметический дефект. E. Jaffe и D. HuLtquist в 1995 г. определили, что «пациенты больше синие, нежели больные». По данным лабораторных исследований выявляют повышение содержания метгемоглобина (15-40%) и количества эритроцитов (компенсаторный эритроцитоз).
Таким образом, анализ данных литературы показывает, что метгемоглобинемия является малоизученным заболеванием и диагноз выставляется достаточно редко, особенно у новорожденных. Представленное ниже клиническое наблюдение вторичной метгемоглобинемии у недоношенного новорожденного — первый случай установленного диагноза в Краснодарском крае.
Клиническое наблюдение
Основной диагноз:
недоношенность 30 нед. Постконцептуальный возраст 39 нед. Врожденная пневмония, тяжелое течение, реконвалесцент.
Осложнения:
бронхолегочная дисплазия, новая форма, тяжелое течение. Полиорганные дисфункции в анамнезе. Перси-стирующая легочная гипертензия в анамнезе. Гипергликемия на фоне приема контринсулярных препаратов (в анамнезе). Некротический энтероколит II стадии по BeLL, реконвалесцент. Гипоксически-геморрагическое поражение центральной нервной системы (ЦНС) в форме периинтравентрикулярных кровоизлияний II степени, ранний восстановительный период, синдром угнетения. Кандидоз, кожные проявления (в анамнезе). Анемия недоношенного смешанного генеза. Ретинопатия недоношенных. Вторичная метгемоглобинемия.
Сопутствующий диагноз:
функционирующее овальное окно. Внутриутробная гипотрофия I степени (ИМТ 12%).
Анамнез
Недоношенный мальчик от первой беременности матери 20 лет, протекавшей на фоне угрозы самопроизвольного выкидыша на 20-й неделе беременности, потребовавшей стационарного лечения. В сроке гестации 24-25 нед беременности диагностирована угроза преждевременных родов на фоне истмико-цервикальной недостаточности, многоводия, установлен акушерский пессарий. Роды первые, оперативные (кесарево сечение с применением обезболивания методом эпидуральной анестезии по общепринятой методике местным анестетиком ропивакаином 0,75%), в сроке гестации 30 нед. Околоплодные воды светлые. Масса тела ребенка при рождении — 1140 г, длина — 34 см, окружность головы — 26 см, окружность груди — 25 см. Оценка по шкале Апгар: 3-4 балла. Масса тела при переводе — 1180 г (+40 г массы тела при рождении).
Развитие заболевания во время нахождения в роддоме: в родильном зале проведена первичная и реанимационная помощь новорожденному — лучистое тепло, продленный вдох, искусственная вентиляция легких (ИВЛ) через лицевую маску в течение 30 с (Peep +5, 21% O2), интубация трахеи, введен порактант альфа в дозе 200 мг/кг, перевод на традиционную ИВЛ.
Ребенок поступил в отделение в тяжелом состоянии, в транспортном инкубаторе на традиционной ИВЛ. Тяжесть состояния была обусловлена дыхательными расстройствами, кислородной зависимостью, неврологической симптоматикой в виде синдрома угнетения ЦНС, морфофункциональной незрелостью на фоне недоношенности.
В условиях родильного дома проводилась терапия: респираторная, антибактериальная — ампициллин/сульбактам 100 мг/кг в сутки, амикацин 15 мг/кг в сутки; энтеральное питание через орогастральный зонд и инфузионная терапия с дотацией частичного парентерального питания и электролитов по физиологической потребности.
На 5-е сутки жизни состояние ребенка оставалось без отчетливой динамики. Недоношенный новорожденный был переведен в отделение реанимации и интенсивной терапии новорожденных № 2 обособленного подразделения Перинатального г. Краснодара для расширения объема диагностического поиска, лечения и выхаживания. Основной диагноз: недоношенность 30 нед. Врожденная пневмония на фоне респираторного дистресс-синдрома новорожденного.
Сопутствующий диагноз: церебральная ишемия II степени, острый период в форме синдрома угнетения ЦНС. Внутриутробная гипотрофия I степени (дефицит массы тела 12%). Функционирующее овальное окно. Открытый артериальный проток.
При поступлении в отделение общее состояние ребенка оставалось тяжелым за счет дыхательной недостаточности, кислородной зависимости, требующей респираторной поддержки, интоксикационного синдрома, неврологической симптоматики в виде синдрома угнетения ЦНС, недоношенности, внутриутробной гипотрофии I степени. Ребенок находился на традиционной ИВЛ (аппаратом «Puritan Bennett» с параметрами: режим А/С; 35 циклов в минуту; Pip +18; Peep +5; Tins — 0,32; FiO2 — 40%).
С момента поступления в отделение в состоянии ребенка обращали на себя внимание бледно-розовые кожные покровы с наличием «серого колорита» с легким цианотичным оттенком, венозный рисунок на коже грудной клетки, акроцианоз, периоральный цианоз, что было расценено как проявление интоксикационного синдрома. В неврологическом статусе отмечались мышечная гипотония, гиподинамия, синдром угнетения. Судорог не зафиксировано. Аускульта-тивно в легких, на фоне аппаратного дыхания, выслушивалось ослабленное дыхание, проводящееся во все отделы, и множественные крепитирующие хрипы во всех точках аускультации. Гемодинамические показатели оставались стабильными. Аускультативно на верхушке сердца систолический шум. Печень +1,5 см ниже края правой реберной дуги, селезенка не пальпировалась.
С целью уточнения диагноза проведен комплекс лабораторных и инструментальных исследований.
1. По данным рентгенографии органов грудной клетки определялись усиление легочного рисунка в прикорневых отделах нижних долей справа и слева и инфильтрация, отсутствовала дифференцировка хвостовой части корней. Отмечалось диффузное усиление легочного рисунка за счет интерстициального компонента. Корни легких были малоструктурны, с нечеткими контурами. Диафрагма расположена обычно. Купол диафрагмы четкий. Сердце не расширено. Аорта без особенностей. Средостение не расширено. Ребра не изменены, межреберья одинаковы с обеих сторон. Заключение: двусторонняя пневмония.
2. Эхо-кардиография: функционирующее овальное окно.
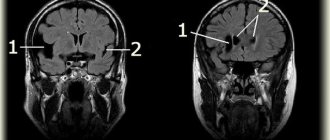
3. Нейросонография: периинтравентрикулярные кровоизлияния II степени. Формирующаяся субэпендимальная киста справа. Диффузное повышение эхогенности перивентрикулярных зон. Асимметричная дилатация боковых желудочков. Индекс резистентности передней мозговой артерии при допплерографии — 0,73.
4. Ультразвуковое исследование почек: диффузное повышение паренхимы обеих почек. Расширение собирательной системы правой почки. Эхопризнаки пиелоэктазии слева. Индекс резистентности почечных артерий при допплерографии — 0,72.
5. Осмотр врачом-неврологом. Диагноз: гипоксически-геморрагическое поражение ЦНС, острый период в форме периинтравентрикулярного кровоизлияния II степени, синдром угнетения.
6. Группа крови и резус ребенка: B (III) Rh (+) положительная. Группа крови и резус матери: O (I) Rh (-) отрицательная.
7. По кислотно-щелочному составу (КЩС): компенсированный метаболический ацидоз, глюкоза — 4,4 ммоль/л, лактат — 2,8 ммоль/л, метгемоглобин — 1,9%.
8. Общий анализ крови без провоспалительных изменений: уровень лейкоцитов 16,7х109/л, палочкоядерные нейтрофилы 9%, сегментоядерные нейтрофилы 33%, гемоглобин — 166 г/л, эритроциты — 4,7х1012/л, тромбоциты -261х109/л. Уровень капиллярной глюкозы 3,8 ммоль/л.
9. По результатам биохимического анализа крови уровень общего билирубина 81,4 мкмоль/л, прямая фракция составила 7,9 мкмоль/л, С-реактивный белок — 0,73 мг/л, трансаминазы в норме, электролитных нарушений не отмечено.
10. Результаты микробиологического исследования крови, трахеи и ануса без роста.
В условиях отделения: продолжена антибактериальная терапия ампициллином + сульбактамом 75 мг/кг в сутки, амикацин 15 мг/кг в сутки; респираторная терапия под контролем газового состава крови; энтеральное питание и частичное парентеральное питание с дотацией белка, жира, электролитов по физиологической потребности; охранительный режим, выхаживание в условиях кувеза, сервоконтроль.
На 8-е сутки жизни состояние ребенка прогрессивно ухудшилась в клинической картине за счет присоединения полиорганных дисфункций в виде нарастания дыхательных расстройств и кислородной зависимости на фоне прогрессирования гипоксии (Sat 86-84%), потребовавших перевода на высокочастотную осцилляторную ИВЛ (ВЧО ИВЛ) со 100% кислородом (по КЩС декомпенсированный метаболический ацидоз, критическая гипоксемия, гиперкапния, уровень метгемоглобина составил 50%), присоединения гемодинамических расстройств, потребовавших подбора доз кардиотонической поддержки со стабилизацией гемодинамики 4% дофамином в дозе максимально 10 мкг/кг в минуту, адреналина 1% 1,0 мкг/кг в минуту, клиническая картина некротического энтероколита II стадии по Bell, выраженного угнетения ЦНС в неврологическом статусе, мышечной гипотонии, анемии тяжелой степени (гемоглобин 111 г/л). Проведена гемотрансфузия эритроцитной массой, обедненной лейкоцитами и тромбоцитами (ЭМОЛТ) 0 (I) Rh отрицательной. На этом фоне цвет кожных покровов с прогрессивно нарастающим выраженным «серым колоритом», появлением центрального цианоза, акроцианоза, цианоза ушных раковин в результате развившейся тяжелой гемической гипоксии. В ОАК: лейкоциты — 14,7х109 /л, гемоглобин — 111 г/л, гематокрит — 35, тромбоциты 274х109 /л, метамиелоциты — 1%, палочкоядерные нейтрофилы — 4%, сегментоядерные нейтрофилы — 46%, лимфоциты — 32%, моноциты — 13%, эозинофилы — 2%, базофилы — 2%.
Учитывая тяжесть состояния в виде присоединения полиорганных дисфункций для исключения реализации внутриутробной инфекции в форме позднего неонатального сепсиса расширен диагностический поиск. Проведено дополнительное обследование ребенка и матери методом парных сывороток для выявления TORCH-комплекса: исследование крови методом иммуноферментного анализа (табл. 1, 2).
У матери и ребенка выявлены положительные иммуноглобулины класса G токсоплазменной инфекции и цито-мегаловируса. При анализе полученных результатов титр материнских антител гораздо выше, чем у ребенка, что свидетельствует о трансплацентарной передаче иммуноглобулинов от матери к плоду (ПЦР-диагностику не проводили).
В качестве пассивной иммунизации к лечению добавлен иммуноглобулин человека нормальный (IgG+IgN+IgA) 5,0 мл/кг в сутки.
На преаналитическом этапе отмечено характерное изменение цвета крови, которая приобрела шоколадно-коричневый цвет.
Учитывая данные литературы [1], повышение фракции метгемоглобина косвенно может являться одним из предикторов септического процесса в организме. Ребенку проводили динамический контроль показателей лейкоцитарной формулы в общем анализе крови.
По результатам исследования общего анализа крови обращало на себя внимание отсутствие роста уровня лейкоцитов в динамике и отсутствие сдвига лейкоцитарной формулы влево, в то время как по данным анализа кислородного статуса, прогрессивно нарастал уровень метгемоглобина, в том числе и в момент ухудшения состояния (рис. 1).
Определяли в динамике уровень СРБ. Он оставался в пределах нормы, что соответствует течению данного заболевания (табл. 3).
При спонтанной манифестации прогрессивный рост метгемоглобина (2-9-18%) на 7-е сутки жизни достиг критического состояния (50%) на 8-е сутки, на фоне нормального уровня лактата по данным кислотно-основного состава крови, с последующим развитием полиорганных дисфункций в виде прогрессирования дыхательной недостаточности и кислородозависимости, потребовавших перевода на ВЧО ИВЛ, нарастания гемодинамических расстройств, неврологической симптоматики в виде синдрома депрессии ЦНС на фоне выраженного центрального цианоза. По линии телемедицины была проведена консультация с гематологом ГБУЗ «Детская краевая клиническая больница» Минздрава Краснодарского края (Краснодар). Ребенку был выставлен диагноз «идиопатическая метгемоглобинемия».
В рамках диагностического поиска полностью исключено наличие у ребенка маркеров системного воспалительного ответа, что подтверждается следующими показателями: отрицательными результатами при анализе уровня прокальцитонина, отсутствием роста патологической микрофлоры из всех локусов организма, нормальным уровнем СРБ (табл. 3) и отсутствием провоспалительных изменений в общем анализе крови (рис. 1). Обследование на TORCH-инфекции (табл. 1, 2) также дало отрицательный результат. По результатам проведенного анализа лабораторных данных, повышение уровня метгемоглобина не коррелировало с динамическими показателями лактата, что, в свою очередь, позволило исключить неонатальный сепсис (рис. 2). При сборе семейного анамнеза не выявлены данные о проявлениях метгемоглобинемии у родителей и ближайших родственников ребенка.
Была добавлена 5% аскорбиновая кислота в дозе 500 мг/кг в сутки. В динамике на фоне проводимой интенсивной терапии с пошаговым снижением дозы 5% аскорбиновой кислоты до 50 мг/кг в сутки уровень метгемоглобина нормализовался до 0,8% к 18-м суткам жизни (см. рис. 1, 2), что послужило поводом к повторной консультации гематолога ГБУЗ «Детская краевая клиническая больница» Минздрава Краснодарского края для решения вопроса об отмене патогенетической терапии. При повышении уровня метгемоглобина в крови до 15-20% в терапию рекомендовано добавить 1% раствор метиленового синего в дозе 1 мг/кг в сутки внутривенно. Для уточнения формы данного заболевания ребенок был консультирован в ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России (Москва). В результате проведенной телемедицинской консультации на основании представленной медицинской документации пациенту рекомендовано определение активности метгемоглобинредуктазы в эритроцитах. С этой целью образцы крови ребенка в объеме 5 мл на антикоагулянте в незамороженном виде при температуре +4…+6 °С были направлены в лабораторию в ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России. Согласно проведенному исследованию в вышеуказанной лаборатории, у ребенка выявлена метгемоглобинемия как состояние, связанное с повышенным содержанием метгемоглобина — 9,3%. Установлено снижение активности НАД-зависимой цитохром-Ь5-редуктазы -1,6 ед. акт. совместно с умеренным снижением коэффициентов Бетке 1,2 (табл. 4).
При проведении электрофореза гемоглобиновых фракций у пациента выявлены аномальные минорные фракции гемоглобина в зоне γ-цепей (зона Z 11 и Е). На основании проведенного специфического лабораторного обследования было дано заключение: врожденная энзимопеническая метгемоглобинемия не подтверждена, ввиду того что данная находка может являться следствием недоношенности ребенка.
Вероятнее всего, у данного пациента метгемоглобинемия носила вторичный (приобретенный) характер, что подтверждают результаты дифференциальной диагностики наследственных метгемоглобинемий (см. табл. 4). С целью исключения медикаментозно вызванной причины развития метгемоглобинемии вторичного характера был проведен подробный анализ лекарственной терапии. Препаратов, содержащих нитро- и аминогруппу, применяемых в терапии у данного ребенка, не выявлено.
За время пребывания в отделении состояние ребенка стабилизировалось. На 30-е сутки жизни он переведен на неинвазивную вентиляцию легких продолжительностью 8 сут с последующим переводом на самостоятельное дыхание с дополнительной подачей увлажненного кислорода через биназальные канюли с концентрацией FiO2 30%. В 2 мес жизни в стабильном состоянии переведен из отделения реанимации и интенсивной терапии № 2 в педиатрическое отделение ГБУЗ «Детская краевая клиническая больница» Минздрава Краснодарского края с целью дальнейшего лечения и выхаживания.
На 74-е сутки жизни ребенок в удовлетворительном состоянии выписан домой под наблюдение отделения катамнеза Детского консультативно-диагностического центра г. Краснодара.
Обсуждение
Метгемоглобинемия является редким заболеванием (по некоторым данным, к настоящему времени в мире описано всего около 600 случаев метгемоглобинемий), клиническая картина которого носит неспецифический характер и может протекать в комплексе с другими симптомами, что затрудняет диагностику. По совокупности ряда симптомов в комплексе с лабораторными методами обследования данный клинический случай представляет актуальную проблему с учетом малого количества информации по данному состоянию в современной неонатологии. Врачам-неонатологам следует быть настороже в отношении данной патологии и иметь возможность своевременной диагностики, направления на консультацию к смежным специалистам с целью уточнения формы данного заболевания, что, возможно, обеспечит благоприятный прогноз.
Конфликт интересов
. Авторы заявляют об отсутствии конфликта интересов.
Литература
1. Казанец Е.Г Метгемоглобинемии // Детская больница. 2009. № 1. С. 38-42.
2. Jaffe E.R. Enzymopenic hereditary methemoglobinemia: a clinical/biochemical classification // Blood Cells. 1986. Vol. 12, N 1. P. 81-90.
3. Казанец Е.Г., Андреева А.П., Хангулов С.В., Токарев Ю.Н. Наследственные цианозы, обусловленные присутствием в крови аномальных гемоглобинов группы М: выявление, идентификация, свойства // Гематол. и трансфузиол. 1990. № 3. С. 9-13.
4. Клейменова И.С., Швырев А.П., Середняк В.Г. Сотникова Н.А. и др. Врожденная энзимопеническая метгемоглобинемия II типа // Рос. вестн. перинатол. и педиатр. 2011. № 6. С. 80-87.
5. Nagai T., Shirabe K., Yubisui T., Takeshita M. Analysis of mutant NADH-cytochrome-b5-reductase: apparent «type III» methemoglobinemia can be explained as type I with an unstable reductase // Blood. 1993. Vol. 81. P. 808-814.
6. Tanishima K., Tanimoto K., Tomoda A. et al. Hereditary methemoglobinemia due to cytochrome-b5-reductase deficiency in blood cells without associated neurologic and mental disorders // Blood. 1985. Vol. 66. P. 1288-1291.
7. Francois J. Cas de cyanose congenitale sans cause apparente // Bull. Acad. Roy Med. Belg. 1845. Vol. 4. P. 698.
8. Hitzenberger K. Autotoxische zyanose (intraglobulare methamoglo-binamie) // Wien. Arch. Intern. Med. 1932. Vol. 23. P. 85-96.
References
1. Kazanets E.G. Methemoglobinemia. Detskaya bol’nitsa [Children’s Hospital]. 2009; (1): 38-42 (in Russian)
2. Jaffe E.R. Enzymopenic hereditary methemoglobinemia: a clinical/ biochemical classification. Blood Cells 1986; 12 (1): 81-90.
3. Kazanets E.G., Andreeva A. P. Khagulov S.V., Tokarev Yu.N. Hereditary cyanosis caused by anomaly hemoglobin group M in blood: revealing, identification, properties. Gematologiya i transfuziologiya . 1990; (3): 9-13. (in Russian)
4. Kleimenova I.S., Shvirev A.P. Serednyak V.G., Sotnikova N.A., et al. Congenital enzymopenic methemoglobinemia of II type. Rossiyskiy vestnik perinatologii i pediatrii . 2011; (6): 80-7. (in Russian)
5. Nagai T., Shirabe K., Yubisui T., Takeshita M. Analysis of mutant NADH-cytochrome-b5-reductase: apparent «type III» methemoglobinemia can be explained as type I with an unstable reductase. Blood. 1993; 81: 808-14.
6. Tanishima K., Tanimoto K., Tomoda A., et al. Hereditary methemoglobinemia due to cytochrome-b5-reductase deficiency in blood cells without associated neurologic and mental disorders. Blood. 1985; 66: 1288-91.
7. Francois J. Cas de cyanose congenitale sans cause apparente. Bull Acad Roy Med Belg. 1845; 4: 698.
8. Hitzenberger K. Autotoxische zyanose (intraglobulare methamoglo-binamie). Wien Arch Intern Med. 1932; 23: 85-96.
Наследственная энзимопеническая
метгемоглобинемия – наследственное заболевание, при котором содержание метгемоглобина (MetHb) в крови превышает физиологическую норму (> 1–2% общего количества Hb) (Казанец Е.Г., 2009). В России наблюдается преимущественно у взрослых в виде эндемических очагов в Якутии в районе реки Вилюй (Токарев Ю.Н. и соавт., 1975, 1976, 1983; Дервиз Г.В., 1977; Нисан Л.Г. и соавт.,1987). Выраженность симптомов обусловлена количеством метгемоглобина в крови. Повышение MetHb до 10% чаще всего не дает клинически выраженных проявлений. При повышении MetHb в пределах 10–20% появляется цианоз слизистых и кожных покровов, возникают общая слабость, недомогание, ослабление памяти, раздражительность, головные боли. При содержании MetHb в пределах 30–50% к вышеперечисленным симптомам присоединяются боли в сердце различного характера, одышка, головокружение, резко выраженный цианоз, повышенная вязкость крови. Содержание MetHb более 70% несовместимо с жизнью.
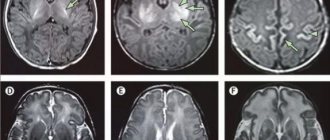
| Симптомы: a) умеренный цианоз губ и слизистой рта, проявления дистонии в мышцах лица; b) цианоз ногтевых пластинок. При метгемоглобинемии 1 типа пациентов периодически беспокоят головные боли, головокружение, одышка, тахикардия, быстрая утомляемость, сонливость, возможно, отставание в физическом и психическом развитии. При метгемоглобинемии 2 типа цианоз сопровождается задержкой интеллектуального развития, прогрессирующей вторичной микроцефалией, нарушением развития нервной системы.МРТ часто обнаруживает корковую и подкорковую атрофию. Лабораторные методы исследования выявляют повышение содержания метгемоглобина (1540%) и количества эритроцитов (компенсаторный эритроцитоз). |
Молекулярно-генетическая причина заболевания
— мутации в гене DIAI, кодирующем две формы фермента NADH-цитохром b5 редуктазы. Мембраносвязанная форма фермента участвует в основных биохимических процессах каждой клетки, растворимая форма участвует в редукции метгемоглобина в эритрорцитах. При мутациях, приводящих к нарушению работы только растворимой формы фермента, возникает 1-ый тип заболевания. При мутациях, нарушающих функционирование обоих форм фермента — второй тип. В России метгемоглобинемия первого типа наиболее часто встречается среди народа саха, частота его в Якутии составляет 1:5700 человек, т.е. каждый 37 якут является гетерозиготным носителем заболевания.Молекулярно-генетической причиной заболевания якутского варианта метгемоглобинемии 1 типа является мутация с.806С›Т в гене DIA1, приводящая к аминокислотной замене Pro269Leu.
Роль красных клеток крови в метаболизме метгемоглобина была установлена Gibson QH в 1943-48 годах. Согласно современным взглядам в эритроцитах одновременно происходит две противоположные реакции, уравновешивающие друг друга. С одной стороны, железо гемоглобина окисляется, превращаясь из двухвалентного в трёхвалентное, при этом образуется не переносящий кислород метгемоглобин (MetHb), за сутки его образуется 0,5-3% от общего количества гемоглобина в организме (Дервиз Г.В., 1977). С другой стороны этот MetHb всё время восстанавливается обратно в функционально активный гемоглобин, в итоге у здоровых людей уровень MetHb удерживается в пределах до 1-1,5%. Процесс восстановления MetHb в эритроцитах изучен довольно хорошо. В эритроцитах известно несколько далеко не равноценных по своей эффективности восстанавливающих систем. 67-73% восстановления MetHb в активный Hb обеспечивает фермент эритроцитов NADH-cytochromeb5 reductase (НАД·Н2-MetHb-редуктаза, NADH-феррицианидредуктаза, NADH-дегидрогеназа, диафораза I, NADH-дегидрогеназа, его роль установлена в 1959 Scott and Griffith). При блокаде этой системы вследствие генетических дефектов стимулируются минорные пути прямого восстановления MetHb эндогенными восстановителями (аскорбиновой кислотой, восстановленным глютатионом, флавином, тетрагидроптерином, цистеином, метаболитами триптофана) или другими системами (Казанец Е.Г., 2009).
Наследственная энзимопеническая метгемоглобинемия (НЭМ), или метгемоглобинемия, обусловленная дефицитом НАДН-цитохром-Ьб-редуетазы (ММ 250800) до сих пор остается мало изученной патологией, особенно у детей (Jaffe E R., 1986; Shirabe К., Yubisui T., 1991; Wu Y., Mota L.V., Kaplan J.C., 1995; Chang-Hui Huang, 1998; Dekker J., Eppink M., 2001). Патогенез клинических проявлений НЭМ определяется хронической гипоксией вследствие окисления части гемоглобина в мет-форму и образования «валентных» гибридов, не способных захватывать кислород в легких и доставлять его тканям (Андреева А.П., 1976, 1977; Захарова Ф.А., 1982; Токарев Ю.Н., 1983). Степень выраженности клинической симптоматики зависит от содержания метгемоглобина в крови и компенсаторных способностей сердечно-сосудистой, дыхательной и гемопоэтической систем в процессе адаптации к гипоксии (Кушаковский М.С., 1968; Ниссан Л.Г., 1987; Аскерова Т.А., 1995).
В литературе имеются описания клинической картины заболевания и состояния периферической крови у взрослых (Захарова Ф.А., 1982; Токарев Ю.Н., 1983; Jenkins J.M., 1992; Shirabe К., 1995; Wang Y., Wu Y., 2000). При этом установлено увеличение содержания гемоглобина, эритроцитов, очевидно, носящее компенсаторный характер (Токарев Ю. Н., 1980, 1983). Захарова Ф.А. (1982) сообщает о повышении уровня сывороточного железа у больных. Однако в литературе нет данных о функционально-морфологическом состоянии периферического звена эритрона при этом заболевании. Имеются лишь единичные сведения об изменениях морфологической характеристики эритроцитов при различных гемоглобинопатиях, сопровождающихся гипоксией (Ковалева Л.Г., Постников Ю.В., 1987; Казанец Е.Г.,1990; Троицкая О.В.,1996, 1999; Nagai Т., 1980).
Клиническая картина наследственной энзиматической метгемоглобинемии у детей имеет возрастные особенности: для школьников характерен более выраженный цианоз, симптомы функциональной кардиопатии и гипоксии. Показатели массы тела и роста у детей с наследственной энзимопенической метгемоглобинемей ниже средних показателей, причем их дефицит увеличивается с возрастом. Периферическое звено эритрона у больных независимо от возраста характеризуется увеличением числа незрелых форм эритроцитов, а также значительным увеличением дегенеративных и плоских форм эритроцитов. Пациенты с наследственной энзимопенической метгемоглобинемей независимо от возраста имели усиление перекисного окисления липидов и снижение активности антиоксидантных систем — каталазы и низкомолекулярных антиоксидантов. Для младшей группы детей характерно увеличение супероксиддисмутазы. Патогенетическая терапия аскорбиновой кислотой приводит к улучшению . состояния детей, снижению уровня метгемоглобина и повышению активности суммарных антиоксидантов.
Течение болезни
, как правило, доброкачественное. Продолжительность жизни пациентов не страдает. Кровь у таких больных темно-коричневого цвета в результате повышения содержания метгемоглобина. В некоторых случаях у нелеченых больных может наблюдаться вторичный компенсаторный эритроцитоз, увеличение содержания гемоглобина (до 170240 г/л), небольшой ретикулоцитоз (менее 3%), повышение вязкости крови и уменьшение СОЭ. Возможно незначительное повышение билирубина в сыворотке крови за счет непрямой (свободной) фракции. У гетерозигот концентрация метгемоглобина в крови составляет 12%, признаки заболевания отсутствуют. Цианоз может появиться после приема метгемоглобинобразующих лекарственных препаратов (Трошин В.А., 2007; Казанец Е.Г., 2009).
В Якутии зарегистрировано необычайно высокое распространение НЭМ I типа у коренных жителей республики (Токарев Ю.Н., 1983, где) частота её составляет 1:5700 человек, т.е. каждый 37 якут является гетерозиготным носителем заболевания. По сути дела, Якутия является единственным очагом данного заболевания на территории России. (За пределами России наследственные доброкачественные метгемоглобинемии распространены среди жителей Гренландии, индейцев Аляски и представителей племени навахос в США).
Возможно, что накопление этого заболевания произошло вследствие эффекта основателя — популяционно-генетического феномена, связанного с ограниченной репродуктивной численностью в прошлом достаточно изолированных популяций (Серошевский В.Л. 1993). Молекулярно-генетическое исследование якутских больных с НЭМ не подтвердило наличие 3 точковых мутаций гена НАДН-цитохром-Ь5-редуктазы (А^5701п, Ьеи72Рго, Уа1105Ме1), встречающихся на территории Азии у японцев и китайцев.
Литература
- Блюменфельд Л.А. Гемоглобин и обратимое присоединение кислорода. М.: Сов. наука, 1957.
- Дервиз Г.В. Наследственная энзимопеническая метгемоглобинемия.//Клиническая медицина, 1977, №5, стр. 8.
- Казанец Е.Г. Метгемоглобинемии.// Детская больница, 2009, №1, стр. 38-42.
- Казанец Е.Г., Андреева А.П., Хангулов С.В., Токарев Ю.Н. Наследственные цианозы, обусловленные присутствием в крови аномальных гемоглобинов группы М: выявление, идентификация, свойства // Гематология и трансфузиология, 1990, №3, с. 9–13.
- Краснопольская К.Д. Наследственные болезни обмена веществ. – М., 2005, 290 с.
- Мельник А.И., Мельник В.А. Обострение наследственной метгемоглобинемии у близнецов грудного возраста//Педиатрия. — 1986. — № 12. — С. 58 — 60.
- Нисан Л.Г., Гуревич С.П., Казанец Е.Г., Фролова М.И., Токарев Ю.Н., Салмова Т.С., Бутина М.В. Наследственная энзимопеническая метгемоглобинемия у новорожденных // Вопросы материнства и детства, 1987, №1, с. 74–75.
- Токарев Ю.Н., Файнштейн Ф.Э. и др.//Проблемы гематологии и трансфузиологии. Т.2. М., 1976, С.123-128.
- Токарев Ю.Н., Холлан С.Р., Корраля-Альмонте Х.С.// Наследственные анемии и гемоглобинопатии. – М., 1983.
- Торшин В. А. Клинически значимые дисгмоглобины. Карбоксигемоглобин // Лаборатория. 2007. № 1. С. 17-18.
- Челноков С.Б., Яковлева Е.А., Пудина Н.А. Случай тяжелой метгемоглобинемии у недоношенного новорожденного ребенка// Вестник интенсивной терапии. 2002. №2. С.1821.
- Aalfs CM, Salieb-Beugelaar GB, Wanders RJ, Mannens MM, Wijburg FA. A case of methemoglobinemia type II due to NADH-cytochrome b5 reductase deficiency: determination of the molecular basis. Hum Mutat 2000;16:18–22.
- Abdemoula MS. La methemoglobinemia hereditaire recessive de type II. A propos d’une observation. Revue Maghrebine de Pediatrie 2002; XII-IV:207–10.
- Bewley MC, Davis CA, Marohnic CC, Taormina D, Barber MJ. The structure of the S127P mutant of cytochrome b5 reductase that causes methemoglobinemia shows the AMP moiety of the flavin occupying the substrate binding site. Biochemistry 2003; 42:13145–51.
- Blumenfeld L.A. Physics of Bioenergetic Processes. N.Y.: Springer-Verlag, 1983.
- Davis CA, Barber MJ. Cytochrome b5 oxidoreductase: expression and characterization of the original familial idiopathic methemoglobinemia mutations E255 and G291D. Arch Biochem Biophys 2004; 425:123–32.
- Dekker J, Eppink MH , van Zwieten R, de Rijk T, Remacha AF, Law LK, et al. Seven new mutations in the nicotinamide adenine dinucleotide reduced-cytochrome b(5) reductase gene leading to methemoglobinemia type I. Blood 2001;97:1106–14.
- Den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000;15:7–12.
- Dickerson R. E., Geis I. Hemoglobin: Structure, Function, Evolution, and Pathology (Benjamin Cummings: Menlo Park, CA), 1983.
- Du M, Shirabe K, Takeshita M. Identification of alternative first exons of NADH-cytochrome b5 reductase gene expressed ubiquitously in human cells. Biochem Biophys Res Commun1997; 235:779–83.
- Ewenczyk C., Leroux A., Roubergue A., Laugel V., Afenjar A., Saudubray J.M., Beauvais P., BillettedeVillemeur T., Vidailhet M., Roze E. Recessive hereditary methaemoglobinaemia, typeII: delineation of the clinical spectrum./ Brain (2008), 131,760-771.
- Fermo E, Bianchi P, Vercellati C, et al. Recessive hereditary methemoglobinemia: Two novel mutations in the NADH-cytochrome b5 reductase gene. /J Blood Cells Mol Dis 2008 Mar 15.
- Fisher RA, Povey S, Bobrow M, Solomon E, Boyd Y, Carritt B. Assignment of the DIA1locus to chromosome 22.Ann Hum Genet 1977;41:151–5.
- Francois. Cas de cyanose conge?nitale sans cause apparente. Bull Acad. Roy Med Belg 1845; 4: 698.
- Fusco C, Soncini G, Frattini D, Giustina ED, Vercellati C, Fermo E, Bianchi P. Cerebellar atrophy in a child with hereditary methemoglobinemia type II./ Brain Dev 2010 Jul 21.
- Gibson QH. The reduction of methaemoglobin in red blood cells and studies on the cause of idiopathic methaemoglobinaemia. Biochem J 1948; 42:13–23.
- Gokalp S, Unuvar E, Oguz F, Kilic A, Sidal M. A case with quadriparetic cerebral palsy and cyanosis: congenital methemoglobinemia. Pediatr Neurol 2005; 33:131–3.
- Gonzalez R, Estrada M, Wade M, de la Torre E, Svarch E, Fernandez O, et al. Heterogeneity of hereditary methaemoglobinaemia: a study of 4 Cuban families with NADH-Methaemoglobin reductase deficiency including a new variant (Santiago de Cuba variant). Scand J Haematol 1978; 20:385–93.
- Grabowska D, Plochocka D, Jablonska-Skwiecinska E, Chelstowska A, Lewandowska I, Staniszewska K, et al. Compound heterozygosity of two missense mutations in the NADH-cytochrome b5 reductase gene of a Polish patient with type I recessive congenital methaemoglobinaemia. Eur J Haematol 2003; 70:404–9.
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18: 2714–23.
- Hegesh E, Hegesh J, Kaftory A. Congenital methemoglobinemia with a deficiency of cytochrome b5. N Engl J Med. 1986; 314:757–761.
- Heusden A, Willems C, Lambotte C,Hainaut H, Chapelle P, Malchair R. . Arch Fr Pediatr 1971; 28: 631–45.
- Higasa K, Manabe JI, Yubisui T,Sumimoto H, Pung-Amritt P, Tanphaichitr VS, et al. Molecular basis of hereditary methaemoglobinaemia, types I and II: two novel mutations in the NADH-cytochrome b5 reductase gene. Br J Haematol 1998; 103: 922–30.
- Hildebrandt A, Estabrook RW. Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Arch Biochem Biophys 1971; 143: 66–79.
- Hirono H. Lipids of liver, kidney, spleen and muscle in a case of generalized deficiency of cytochrome b5 reductase in congenital methemoglobinemia with mental retardation. Lipids 1984; 19: 60–3.
- Hitzenberger K. Autotoxische zyanose (intraglobulare methamoglobinamie). Wien Arch Intern Med.1932;23:85.
- Hultquist DE, Passon PG. Catalysis of methaemoglobin reduction by erythrocyte cytochrome b5 and cytochrome b5 reductase. Nature1971; 229: 252–4.
- Jablonska-Skwiecinska E, Holtorp-Tyszkiewiczowa J, Staniszewska K. Generalized deficiency of the NADH-methemoglobin reductase in congenital methemoglobinemia with neurological symptoms. Biomed Biochim Acta 1984; 43: S98–100.
- Jaffe ER, Hultquist DE. Cytochrome b5 reductase deficiency and Enzymopenic hereditary methemoglobinemia. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Basis of Inherited Disease.7th ed. NewYork, NY: McGraw-Hill;1995:2267–2280.
- Junien C, Leroux A, Lostanlen D, Reghis A, Boue J, Nicolas H, et al. Prenatal diagnosis of congenital Enzymopenic methaemoglobinaemia with mental retardation due to generalized cytochrome b5 reductase deficiency: firstre port of two cases. Prenat Diagn 1981; 1: 17–24.
- Kaftory A, Freundlich E, Manaster J, Shukri A, Hegesh E. Prenatal Diagnosis of congenital methemoglobinemia with mental retardation. Isr J Med Sci 1986; 22: 837–40.
- Kobayashi Y, Fukumaki Y, Yubisui T, Inoue J, Sakaki Y. Serine-proline replacement at residue 127 of NADH-cytochrome b5 reductase causes hereditary methemoglobinemia, generalized type. Blood 1990; 75:1408–13.
- Kugler W, Pekrun A, Laspe P, Erdlenbruch B, Lakomek M. Molecular basis of recessive congenital methemoglobinemia, types I and II: Exon skipping and three novel missense mutations in the NADH-cytochrome b5 reductase (diaphorase 1) gene. Hum Mutat 2001; 17: 348.
- Lamy M, Frezal J, Jammet ML, Josso N. . Nouv Rev Fr Hematol 1963; 3: 105–20.
- Leroux A, Mota Vieira L, Kahn A. Transcriptional and translational mechanisms of cytochrome b5 reductase isoenzyme generation in humans. Biochem J 2001; 355: 529–35.
- Lostanlen D, Vieira de Barros A, Leroux A, Kaplan JC. Soluble NADH-cytochrome b5 reductase from rabbit liver cytosol: partial purification and characterization. Biochim Biophys Acta 1978; 526: 42–51.
- Manabe J, Arya R, Sumimoto H, Yubisui T, Bellingham AJ, Layton DM, et al. Two novel mutations in the reduced nicotinamide adenine dinucleotide (NADH)-cytochrome b5 reductase gene of a patient with generalized type, hereditary methemoglobinemia. Blood 1996; 88: 3 208 – 15.
- Maran J, Guan Y, Ou CN, Prchal JT. Heterogeneity of the molecular biology of methemoglobinemia: a study of eight consecutive patients. Haematologica 2005; 90: 687–9.
- Nussenzveig RH, Lingam HB, Gaikwad A, Zhu Q, Jing N, Prchal JT. A novel mutation of the cytochrome-b5 reductas gene in an Indian patient: the molecular basis of type I methemoglobinemia. Haematologica 2006; 91: 1542–5.
- Orsini A, Vovan L, Brusquet Y, Gabriel B, Sebag F, Galtier M. Congenital methemoglobinemia due to NADH (DPNH) dependent methemoglobin reductase deficiency. Mars Med 1972; 109: 279–81.
- Owen EP, Berens J, Marinaki AM, Ipp H, Harley EH. Recessive congenital methaemoglobinaemia type II a new mutation which causes incorrect splicing in the NADH-cytochrome b5 reductase gene. J Inherit Metab Dis 1997; 20: 610.
- Percy MJ, Crowley LJ, Davis CA, McMullin MF, Savage G, Hughes J, et al. Recessive congenital methaemoglobinaemia: functional characterization of the novel D239G mutation in the NADH-binding lobe of cytochrome b5 reductase. Br J Haematol 2005; 129: 847–53.
- Ronconi G, Ferracin G. . Riv Clin Pediatr 1964; 74: 152–9.
- Roussel A, Maestraggi P, Tremoulet M, Marchand. A new case of recessive congenital methemoglobinemia. Arch Fr Pediatr 1963; 20: 745–50.
- Sacerdotti-Favini. Methemoglobinemia costituzionale con cerebropatia e oligofrenia. Acta pediat Lat 1948; 11: 255.
- Scott EM, Griffith IV. The enzyme defect of hereditary methemoglobinemia: diaphorase. Biochim Biophys Acta.1959;34:584–586.
- Shirabe K, Landi MT, Takeshita M, Uziel G, Fedrizzi E, Borgese N. A novel point mutation in a 3’splice site of the NADH-cytochrome b5 reductase gene results in immunologically undetectable enzyme and impaired NADH-dependent ascorbate regeneration in cultured fibro-blasts of a patient with type II hereditary methemoglobinemia. Am J Hum Genet 1995; 57: 302–10.
- Shonola S. Da-Silva, Imran S. Sajan and Joseph P. Underwood, III. Congenital Methemoglobinemia: A Rare Cause of Cyanosis in the NewbornA Case Report./ Pediatrics, 2003;112;e158-e161.
- Shotelersuk V, Tosukhowong P, Chotivitayatarakorn P, Pongpunlert W. A Thai boy with hereditary Enzymopenic methemoglobinemia type II. J Med Assoc Thai 2000; 83: 1380–6.
- Smith, R. P. Chapter 11, Toxic responses of the blood. In Casarett and Doull’s Toxicology. The Basic Science of Poisons, 5th ed., C. D. Klaassen, Ed., McGraw-Hill, New York, 1996.
- Strittmatter P, Spatz L, Corcoran D, Rogers MJ , Setlow B, Redline R. Purification and properties of rat liver microsomal stearyl coenzyme A desaturase. Proc Natl Acad Sci USA 1974; 71: 4565–9.
- Takeshita M, Matsuki T, Tanishima K, Yubisui T, Yoneyama Y, Kurata K, et al. Alteration of NADH-diaphorase and cytochrome b5 reductase activities of erythrocytes, platelets, and leukocytes in hereditary methaemoglobinaemia with and without mental retardation. J Med Genet 1982; 19: 204–9.
- Toelle SP, Boltshauser E, Mossner E, Zurbriggen K, Eber S. Sever neurological impairment in hereditary methaemoglobinaemia type 2. Eur J Pediatr 2004; 163: 207–9.
- Trost C. The blue people of Troublesome Creek. Science 82, November, pp. 35-39, 1982.
- Vieira M, Kaplan JC, Kahn A, Leroux A. Four new mutations in the NADH-cytochrome b5 reductase gene from patients with recessive congenital methemoglobinemia type II. Blood 1995; 85: 2254–62.
- Wang Y, Wu YS, Zheng PZ, Yang WX, Fang GA, Tang YC, et al. A novel mutation in the NADH-cytochrome b5 reductase gene of a Chinese patient with recessive congenital methemoglobinemia. Blood 2000; 95: 3250–5.
- Williamson DA, Black JA. Congenital methaemoglobinaemia; a case report. Great Ormond St J 1954; 7: 56–61.
- Wu YS, Wang Y, Huang CH, Lan FH, Zhu ZY. A compound heterozygote in the NADH-cytochrome b5 reductase gene from a Chinese patient with hereditary methemoglobinemia type I. Int J Hematol 2000;72: 34–6.
- Yawata Y, Ding L, Tanishima K, Tomoda A. New variant of cytochrome b5 reductase deficiency (b5RKurashiki) in red cells, platelets, lymphocytes, and cultured fibroblasts with congenital methemoglobinemia, mental and neurological retardation, and skeletal anomalies. Am J Hematol 1992; 40: 299–305.
- Yilmaz D, Cogulu O, Ozkinay F, Kavakli K, Roos D. A novel mutation in the DIA1 gene in a patient with methemoglobinemia type II. Am J Med Genet A 2005; 133: 101–2.
- Yuksel D, Senbil N, Yilmaz D, Yarali N, Gurer YK A rare cause of mental motor retardation: recessive congenital methemoglobinemia type II. /Turk J Pediatr 2009 Mar-Apr; 51(2):187-9.
- Zorc J, Kanic Z. A cyanotic infant: true blue or otherwise? Pediatr Ann.2001;30:597–601.
Метгемоглобинемия
Метгемоглобинемия — состояние, характеризующееся повышенным содержанием метгемоглобина (окисленного гемоглобина) в крови и тканевой гипоксией. Развитие метгемоглобинемии сопровождается акроцианозом, слабостью, головными болями, головокружением, сердцебиением, одышкой при нагрузке. Характерным признаком метгемоглобинемии служит коричнево-шоколадный цвет крови. Для подтверждения диагноза проводится оценка симптоматики, лабораторные исследования и тесты. При тяжелой степени метгемоглобинемии показана кислородотерапия, введение аскорбиновой кислоты, раствора метиленового синего, в ряде случаев — обменная гемотрансфузия. Симптомы метгемоглобинемии
Признаки наследственной метгемоглобинемии становятся заметны в период новорожденности. На коже и видимых слизистых ребенка (в области губ, носогубного треугольника, мочек ушей, ногтевого ложа) заметен цианоз. Кроме наследственной метгемоглобинемии, у детей часто выявляются другие врожденные аномалии — изменения конфигурации черепа, недоразвитие верхних конечностей, атрезия влагалища, талассемия и пр. Нередко дети отстают в психомоторном развитии. В зависимости от уровня фракции MtHb, выраженность проявлений врожденной и приобретенной метгемоглобинемии может значительно варьировать.
При концентрации MtHb в крови:
- 3-15% — кожные покровы приобретают сероватый оттенок
- 15-30% — развивается цианоз, кровь становится шоколадно-коричневого цвета
- 30-50% — появляется слабость, головная боль, тахикардия, одышка при физической нагрузке, головокружение, возникают обмороки
- 50-70% — возникает аритмия, учащенное дыхание, судороги; развивается метаболический ацидоз; отмечаются признаки угнетения ЦНС, возможна кома
- >70% — выраженная гипоксия, летальный исход.
Для любых форм метгемоглобинемии характерна грифельно-серая окраска кожных покровов, однако отсутствуют характерные для сердечно-легочных заболеваний изменения ногтевых фаланг по типу ‘барабанных палочек’. Акроцианоз усиливается при охлаждении, употреблении в пищу нитратосодержащих продуктов, при токсикозах беременности у женщин, а также приеме метгемоглобинобразующих медикаментов.
Лечение и профилактика метгемоглобинемии
Больные с отсутствием клинических проявлений не нуждаются в специальной терапии. При значительной концентрации MtHb в крови и развернутой симптоматике метгемоглобинемии назначается медикаментозная терапия, способствующая превращению метгемоглобина в гемоглобин. Такими восстанавливающими свойствами обладают аскорбиновая кислота и метиленовый синий (хромосмон). Аскорбиновая кислота назначается внутрь сначала в больших, а по мере нормализации состояния — в поддерживающих дозах. Раствор метиленовой сини вводится внутривенно. При выраженном цианозе проводится кислородная терапия. Тяжелая форма метгемоглобинемии является показанием к обменному переливанию крови. Течение наследственной и лекарственной метгемоглобинемии, как правило, доброкачественное. Неблагоприятный исход возможен при тяжелых формах токсической метгемоглобинемии с высоким содержанием MtHb в эритроцитах. Пациентам с подобной патологией следует избегать контакта с метгемоглобинобразующими веществами, переохлаждений и других провоцирующих факторов. Профилактика врожденной метгемоглобинемии заключается в проведении медико-генетической консультации для выявления гетерозиготных носителей среди будущих родителей.