Platelet dysfunction
Specific platelet function disorders
Abnormalities in platelet function
Platelet receptor defects
Glanzmann's thrombasthenia
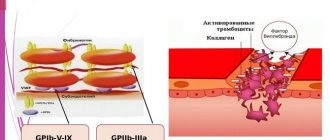
Glanzmann's thrombasthenia (TG) is a disorder of platelet function caused by the absence or reduction of platelet receptor IIb-IIIa. The platelet count, size, shape, and lifespan for this disease are within normal limits. Glanzmann's thrombasthenia is inherited in an autosomal recessive manner: i.e. In parents, this disease is not detected during their lifetime. Glanzmann's thrombasthenia is observed equally in men and women. Bleeding time with TG is always prolonged. Thrombus formation is incomplete or absent. The platelet reaction occurs only with ristocetin. Adhesion in the area of damaged endothelium is normal, but the recruitment of additional platelets into the primary thrombus does not solve the problem. Studying the function of platelet receptors GP IIb-IIa is possible only in special laboratories. Desmopressin is not effective in this case, so platelet mass therapy is performed. Reactions to multiple platelet injections can be quite severe. Therefore, transfusions should be performed with caution.
Bernard-Soulier syndrome
Bernard-Soulier syndrome is a fairly rare disease characterized by an increase in platelet size and a slight decrease in their number. Bleeding time is noticeably prolonged. A study of platelet aggregation in this case shows that the reaction with ristocetin is abnormal. This disorder occurs due to the absence or defectiveness of platelet Ib-IX receptors and von Willebrand factor receptors. This disease must be distinguished from von Willebrand disease, which is caused by a defect in von Willebrand factor rather than in platelet receptors. Bernard-Soulier syndrome is inherited in an autosomal recessive manner: i.e. In parents, this disease is not detected during their lifetime. Bernard-Soulier syndrome is observed equally in men and women. Von Willebrand disease is inherited in a dominant-recessive manner. In the treatment of Bernard-Soulier syndrome, platelet transfusions are performed; however, as with Glanzmann's thrombasthenia, patient alloimmunization may occur.
Defects in granule contents and storage deficiencies
Storage deficiencies are a heterogeneous group of diseases characterized by the inability of platelets to store special substances in cell granules.
Storage deficiencies not associated with systemic diseases Gray platelet syndrome Gray platelet syndrome is a disease caused by a lack of protein in the contents of alpha granules of platelets and megakaryocytes. These proteins include: platelet factor 4, beta thromboglobin, fibrinogen and intracellular growth factor. In a peripheral blood sample, platelets are gray and unnaturally large in size. Aggregation with thrombin is impaired. The patient should undergo a trial of desmopressin before receiving a platelet transfusion. Platelet transfusions may be required if major bleeding occurs or if there is resistance to desmopressin.
Quebec platelet syndrome
Quebec syndrome is an autosomal dominantly inherited disease in which there is a significant impairment of platelet aggregation when tested with epinephrine. This is due to a defect in the proteolysis of the protein in alpha granules and a deficiency of alpha granule multimerin, a protein that binds factor V in the granule, and thus causes a decrease in factor V and a number of other proteins (fibrinogen, von Willebrand factor, etc.)
Storage deficiencies associated with other systemic diseases Hermanski-Pudlak syndrome Hermanski-Pudlak syndrome is inherited in an autosomal recessive manner. Along with it, patients have cutaneous albinism and ceroid-like pigment in bone marrow macrophages. With this syndrome there is also a prolongation of bleeding time and a slight violation of blood clotting. Solid platelet granules are defective. Platelet function studies reveal abnormal aggregation in reaction with ADP, epinephrine, ristocytin and collagen.
Chediak-Higashi syndrome
Chediak-Higashi syndrome is a rare autosomal recessive disorder characterized by abnormally large platelet granules that are similar in size to those of melanocytes, leukocytes, and fibroblasts. In Chediak-Higashi syndrome, patients have partial albinism and recurrent pyogenic infection. The platelet count is within normal limits, bleeding time is prolonged, hard granules are reduced, platelet aggregation is impaired, causing a tendency to bleed.
Wiskott-Aldrich syndrome
Wiskott-Aldrich syndrome is a rare disease, transmitted in a recessive manner, linked to the X chromosome. It is characterized by a decrease in platelet size and thrombocytopenia. Patients with this disease suffer from bleeding caused by impaired platelet count and function. Some also have a storage disorder. Patients with Wiskott-Aldrich syndrome also suffer from recurrent infections and eczema. Laboratory tests indicate the absence of isohemaglutanins. In parallel, immunological disorders are also present. Genetic analysis reveals pathology in many patients. Treatment of acute bleeding is carried out by transfusion of platelet mass. Bone marrow transplantation may be considered as a last resort in the treatment of these patients.
Release defects
Release defects represent the largest group of platelet dysfunctions. Release defects are a heterogeneous group of diseases with a large number of underlying disorders whose mechanism of action is not yet fully understood. Their cause may be a violation of signal transmission from the membrane, abnormal intracellular metabolism, or an anomaly in the release mechanism caused by defects in the structural elements of its participants. A common pathology in this group of diseases is the inability of platelets to successfully release granule contents upon platelet activation. Release defects are accompanied by prolongation of bleeding time and impaired in-vitro platelet aggregation in the presence of ADP, epinephrine and collagen. With more in-depth studies, a defect in ADP release is noted. At the same time, the reserves of ADP in the body, not bound in granules, are not reduced. The contents of the granules are normal. Many patients with release defects respond to treatment with DDAVP, a synthetic analogue of vasopressin.
Application of DDAVP
DDAVP, at a concentration of 4 micrograms/mL, can be used at a dose of 0.3 micrograms/kg of patient body weight intravenously or subcutaneously. There is also an intranasal form of DDAVP. Intranasal DDAVP, used to treat release defects, is more concentrated (1.5 milligrams/ml) than the version used to treat diabetes and enuresis. DDAVP should be used with caution as it has a strong antidiuretic effect and can cause water retention and hyponatremia. These side effects are more severe in children under three years of age and in patients undergoing surgery that requires significant intravenous infusions. If possible, repeat DDAVP injections within a short period of time should be avoided. If this cannot be avoided, it is necessary to introduce electrolytes into the blood serum. Despite this shortcoming, DDAVP is an important step forward, since more than 50% of diseases associated with release defects are susceptible to this type of treatment.
Defects in plasma factors affecting platelet function Defects in plasma factors can lead to impaired platelet function, despite a normal platelet count and intact platelet function. The most common disease in this category is von Willebrand disease. The absence of plasma or platelet fibrinogen leads to defects in platelet function, since fibrinogen is an important factor in platelet-platelet interaction during the formation of the primary clot. Afibrinogenemia is a rare autosomal recessive disorder. Von Willebrand's disease and afibrinogenemia cause platelet adhesion disorders: VWD in the interaction of platelets and the vascular wall, and afibrinogenemia in the interaction of platelets with each other.
von Willebrand disease
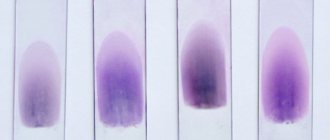
In von Willebrand disease (VWD), the function of von Willebrand factor (vWF), which is of great importance in the functioning of platelets, whose platelet receptor is Ib-IX, is impaired. VWF defects are expressed in subcutaneous hemorrhages and bleeding from the mucous membranes. BV is inherited in an autosomal dominant manner and occurs in both men and women. The bleeding time in this disease is usually prolonged. Various types of von Willebrand disease are associated with disturbances in factor VIII activity, the amount of vWF antigen, and vWF activity, as determined by the restocetin cofactor test. The structure of the protein itself is examined by gel electrophoresis of the multimer.
Afibrinogenimia
This rare disease with an autosomal recessive inheritance is associated with a significant decrease or complete absence of fibrinogen. Bleeding time in this case is usually prolonged. Some patients also experience a decrease in the number of platelets and impaired platelet aggregation. The absence or significant deficiency of fibrinogen in the blood plasma leads to inadequate interaction of platelets with each other.
Defects in the interaction of platelets with plasma clotting factors caused by a defect in platelet coagulant activity Scott syndrome Scott syndrome is the best known in this category of platelet function disorders. In Scott syndrome, platelets form defective complexes of factors Va-X and VIIIa-Ixa. Defects in the binding of these plasma coagulation factor complexes lead to incomplete activation of factor X and prothrombin, as well as disturbances in the activity of platelet factor 3. The reason for this is a violation of the production of phosphatidylserine in the plasma membrane.
Various congenital diseases Disorders of platelet function associated with connective tissue diseases are known. These diseases include: Ehlers-Danlos syndrome, Marfan syndrome, idiopathic osteopsatirrosis and fragility X syndrome. May-Heggelin anomaly is an autosomal dominant disease characterized by ineffective thrombocytopoiesis with normal platelet function, and granule deficiencies in leukocytes. There is also a dysfunction of platelets in Down syndrome. Thrombocytopenia and radial bone defect syndrome are often accompanied by thrombocytopathy. Children suffering from this disease may experience severe hemorrhages in the first year of life, sometimes with fatal consequences. But subsequently thrombocytopenia gradually disappears. This group of patients is indicated for prophylactic platelet transfusion. Hereditary idiopathic thrombocytopenic purpura is also often associated with thrombocytopathy. A connection between platelet function disorders and increased levels of serum immunoglobulin, nephritis, and deafness has also been noted.
Conclusion
Thrombocytopathies constitute a large, heterogeneous group of bleeding disorders ranging in severity from mild to severe. The disease may be asymptomatic; however, most diagnosed patients have its manifestations in the form of petechization and bleeding from the mucous membranes, as well as after injury or surgery. The complex biochemical mechanism of signal transduction requires further understanding. Despite significant advances in the study of the etiology of these phenomena, methods of their treatment remain at a low level. For platelet function disorders associated with defects in plasma coagulation proteins, such as von Willebrand disease and afibrinogenemia, treatment consists of replacement therapy with concentrates of the missing factors. Additional therapy (antifibrinolytics, fibrin glue, etc.), desmopressin and platelet transfusions are the main ones today. Corticosteroids may play a significant role in the treatment of some of these diseases and lead to a reduction in bleeding time, especially with storage defects in patients not responsive to DDAVP.
Platelet hemostasis
Study of platelet aggregation ability
Evaluation of patients with bleeding requires a thorough history, including family history, medication history, physical examination, and only then laboratory testing.
Quantitative and functional platelet abnormalities are common in patients with bleeding disorders.
Types of platelet dysfunction:
| Platelet dysfunction | Probable diagnosis |
| Impaired platelet adhesive function | Bernard-Soulier syndrome Velocardiofascial syndrome Glanzmann's thrombasthenia Congenital afibrinogenemia Von Willebrand disease (possibly a platelet type of von Willebrand disease) Collagen receptor defects |
| Platelet receptor abnormalities | TxA2 receptor defects α-adrenergic receptor defect P2Y12 receptor defect Antiplatelet drugs (eg, clopidogrel) |
| Platelet granule abnormalities | Deficiency of dense granules - deficiency of α-granules - gray platelet syndrome |
| Abnormalities in secretion of the contents of platelet granules, or disruption of signal transduction in platelets | Primary secretion defects Aggregation defects that are similar to those seen in platelet pool storage disorders, but with normal granule and TxA2 content Defects in platelet surface receptor agonists - epinephrine - thromboxane - ADP - collagen Defects in protein G activation (defects in intracellular signal transduction) |
| Arachidonic acid metabolism disorder | Cyclooxygenase defects Thromboxane synthase deficiency Taking drugs |
| Disturbance of the procoagulant mechanism of platelets | Scott syndrome |
| Platelet cytoskeleton defects | MYH9-associated disorders Wiskott-Aldrich syndrome |
Before performing specific tests examining the functional properties of platelets, it is recommended to perform the following laboratory tests:
- Complete blood count with determination of platelet count and average platelet volume. This is important for hereditary macrothrombocytopenia (Bernard-Soulier syndrome), microthrombocytopenia (Wiskott-Aldrich syndrome), and for some bone marrow diseases.
- Study of blood smear and platelet morphology. Necessary for establishing diagnoses such as pseudothrombocytopenia, gray platelet syndrome. In some cases, for example, with Hegglin's thrombocytopenia, a blood smear may show the presence of Dele bodies (light gray-blue oval basophilic leukocyte inclusions located in the peripheral cytoplasm of neutrophils).
Specific laboratory tests to determine the functional properties of platelets include examinations such as:
- screening tests reflecting the functional activity of platelets, for example, activated clotting time (ACT), bleeding time (BT) and PFA-100, - aggregometry, for example, classical Born aggregometry , - flow cytometry to quantify the presence or absence of platelet membrane glycoproteins, — specialized studies used in research laboratories.
The principle of the Born aggregometry method:
The method for measuring platelet aggregation using the Born turbidimetric method is based on recording changes in light transmission in a platelet suspension. These changes are due to a decrease in light scattering and an increase in light transmission (transparency) of the suspension during the formation of platelet aggregates.
Classical Born aggregometry uses platelet-rich plasma (PRP). In the aggregometer, BTP is mixed in a cuvette at a temperature of 37°C. When an agonist is added, the resulting platelet aggregates absorb less light, which is detected by a photocell.
PRP – platelet-poor plasma, PRP – platelet-rich plasma.
The principles of Born aggregometry can be clearly seen in the animated English-language presentation, kindly provided by the authors of the scientific website https://www.platelet-research.org - https://www.platelet-research.org/3/aggregometry.htm.
Pre-analytical variability
Drugs that block platelet function include NSAIDs (non-steroidal anti-inflammatory drugs), specific antiplatelet drugs such as clopidogrel, as well as some antibiotics, antidepressants, beta blockers, etc. Foods such as garlic, turmeric can also affect platelet function and caffeine, high-fat foods (these can cause chylomicrons in the plasma, which interferes with normal light transmission during platelet aggregation testing). A number of factors can influence the accuracy of platelet aggregation results:
- very high or low platelet count;
- temperature - blood samples for platelet aggregation testing should be stored at room temperature;
— pH — platelet aggregation should be carried out at a physiological pH value;
- fibrinogen concentration.
Platelet agonists:
The addition of a platelet agonist to PRP leads to activation of platelets, a change in their shape from discoid to spherical, which is associated with an increase in optical density during platelet activation. The only exceptions to this are adrenaline (epinephrine), which does not change the shape of platelets, and ristocetin, which causes platelet agglutination rather than aggregation.
There are two types of agonists:
- strong agonists - collagen, thrombin, ThA2, arachidonic acid - directly cause platelet aggregation, synthesis and secretion of ThA2 platelet granules.
- weak agonists - ADP and adrenaline - cause platelet aggregation without causing secretion.
Strong agonists at low concentrations may act as weak agonists, but weak agonists, even at high concentrations, will not act as strong agonists.
With some weak agonists (ADP and epinephrine), the platelet aggregation curve may have a biphasic appearance - an initial wave of aggregation (primary wave), followed by a secondary wave of aggregation, which is usually irreversible. At higher ADP concentrations, the two waves of aggregation merge and there is no biphasic signal.
The aggregation response to the agonist is enhanced by the release of TxA2 from membrane phospholipids and the secretion of ADP from dense granules. ADP and TxA2 are agonists that, interacting with their specific receptors, enhance the platelet aggregation reaction.
Working concentrations of the agonists used and their mechanism of action:
| Agonist | Working concentration (C) | Mechanism of action |
| ADF | Low –1.0 μM, 2.5 μM, 5.0 μM High – 10.0 μM | ADP binds to its receptor on the surface of platelets. Initial binding results in the release of intracellular calcium and a change in platelet shape, leading to the formation of a primary wave of aggregation. The secondary wave reflects the release of ADP from platelet storage granules. A low dose of ADP causes only primary aggregation and its effect is reversible. ADP binds to two G-protein receptors P2Y1 and P2Y12. Binding of ADP to the P2Y1 receptor causes a shape change and initiates platelet aggregation (primary wave) by mobilizing calcium. The P2Y12 receptor is considered the main ADP receptor and is responsible for complete platelet aggregation through inhibition of adenyl cyclase. The P2Y12 receptor is also a target of clopidogrel. The second wave of ADP-induced aggregation is suppressed by aspirin and NSAIDs. |
| Adrenalin | 5μM, 10μM | Epinephrine binds to the α2-adrenergic receptor on the surface of platelets, resulting in inhibition of adenyl cyclase and the release of calcium ions. Platelet aggregation with epinephrine is visually similar to aggregation with ADP, with an initial primary wave of aggregation, release of stored ADP from platelets, and a second wave of irreversible aggregation. As with ADP, this second wave of aggregation is inhibited by aspirin and NSAIDs. Adrenaline, like ADP, is considered a weak agonist. |
| Ristocetin | Low –0.5 mg/mL High –1.5 mg/mL,5 mg/mL | Ristocetin in high concentrations causes platelet agglutination through von Willebrand factor and the GPIb-IX-V complex. |
| Collagen | 1.4μg/mL | Collagen binds to the GPVI and GPIa/IIa receptors, inducing the release of granule contents, the TXA2 pool, and then leads to sustained activation of GPIIb-IIIa. The GPIa/IIa receptor is involved in platelet adhesion. The GPVI receptor is involved in platelet signaling and TxA2 generation. The lag phase is formed after the addition of collagen to PRP, usually in less than 1 min. |
| Arachidonic acid | 500μg/mL | Arachidonic acid is a precursor of TxA2 in platelets. Arachidonic acid is converted to TxA2 with the participation of cyclooxygenase and thromboxane synthetase. TxA2 is a potent inducer of platelet aggregation, causing the release of granules, the formation of even greater generation of TxA2 and then the sustained activation of GPIIb-IIIa receptors. |
| Thrombin | Low –50 nmol/L High – 100 nmol/L | Thrombin is the most potent physiological platelet activator, acting on the PAR1 and PAR4 receptors. Thrombin causes platelet aggregation even at low concentrations, this does not lead to complete aggregation, but it changes the shape of platelets. At higher concentrations, thrombin induces complete platelet aggregation. |
Abbreviations used: PRP - platelet-poor plasma, PRP - platelet-rich plasma, TxA2 - thromboxane A2, C - concentration, GP - glycoprotein.
View of the classic two-phase platelet aggregation curve:
With the introduction of adrenaline and low doses of ADP, a biphasic aggregation curve is usually formed, while with the introduction of other agonists, or at high concentrations of inducers, a merger of the first and second waves is possible. In biphasic aggregation, the second wave is associated with the release reaction of platelet granules.
Aggregation parameters.
The main parameters of the aggregogram can be interpreted only after a preliminary assessment of platelet activity according to the types of aggregator curves (reversible, biphasic, irreversible and no platelet aggregation).
The main parameters of the aggregogram are the intensity of primary ( Tpa ), secondary ( Twa ), maximum aggregation ( Tma ) and disaggregation ( Tda ). The dynamics of the aggregation process on the graph should be judged by the slope angles of the aggregogram curve at the stage of primary aggregation ( α1 ), secondary aggregation ( α2 ) and disaggregation (β ). A characteristic of the dynamics of the reaction of release of endogenous stimulators of platelet aggregation is the latent period for collagen-induced aggregation ( tlp ) and the time of onset of secondary aggregation ( twa ) is normal for biphasic aggregation curves (high doses of ADP and adrenaline).
For collagen-induced aggregation, it is also important to evaluate the shortening or lengthening of the latent period ( tlp ) compared to the value of this indicator in healthy people.
A qualitative assessment of the aggregogram allows not only to diagnose hyper- or hyporeactivity of platelets, but also to suggest the causes of their occurrence. To quantitatively register various types of aggregation diagrams, the following indicators should be calculated:
- with irreversible aggregation - Tma, Tpa, Tda, Tba, α1, α2, of which the most informative are Tba, α2 and Tba;
- with reversible aggregation - Tma and Tda.
Fluctuation range of the main indicators of the aggregation diagram:
| Aggregogram parameters | Range of changes | Aggregogram parameters | Range of changes |
| Tma | 30-50% | Tva | 30-45 s |
| Twa | 30-40% | α1 | 60-75º |
| Tda | 60-80% (of Tpa value) | α2 | 60-75º |
| Tpa | 10-20% | β | 30-45º |
| tlp | 25-30 s | ||
Reference values
Normal aggregation activity when using inductors is:
- Platelet aggregation with ADP (5.0 µmol/ml): 60-90%
- Platelet aggregation with ADP (0.5 µmol/ml): 1.4-4.3%
- Platelet aggregation with adrenaline: 40-70%
- Platelet aggregation with collagen: 50-80%
Data interpretation
| Tentative diagnosis | Changes in the aggregogram |
| Glanzmann's thrombasthenia or afibrinogenemia | Absence or significant decrease in aggregation upon stimulation with any agonists except ristocetin |
| Bernard-Soulier syndrome or von Willebrand disease | Absence or significant reduction of aggregation with ristocetin |
| Storage pool violation | Primary wave of aggregation with only ADP, adrenaline and collagen. |
| Action of aspirin or aspirin-like effect | No aggregation with arachidonic acid. Presence of a primary wave only with ADP. Decreased or absent aggregation with collagen. |
| Effect of clopidogrel | No aggregation with ADP |
| Type 2B von Willebrand disease (VWD) or platelet type VWD | Aggregation with low dose of ristocetin (0.5 mg/ml) |
Link to the manual “Clinical and laboratory diagnosis of platelet function disorders”. Vasiliev S.A., Berkovsky A.L., Melkumyan A.L., Suvorov A.V., Mazurov A.V., Kozlov A.A. Federal State Budgetary Institution Hematological Research Center, Ministry of Health of the Russian Federation, Moscow, 2013.
When preparing this section, data from the scientific website www.practical-hemostasis.com was used.