Coarctation of the aorta
Like the patent ductus arteriosus, coarctation of the aorta, strictly speaking, is not a defect of the heart itself, since it is located outside of it and affects the vessels, or rather, the aorta. The heart itself is normal. However, coarctation is always classified as a heart defect, since it affects the entire circulatory system, and the formation of coarctation itself is directly related to cardiac structures and is often combined with other congenital heart defects.
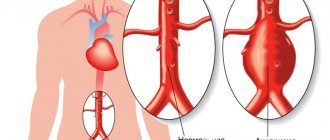
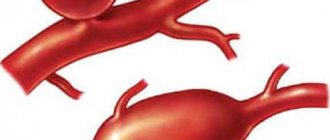
The word coarctation means narrowing. In typical, most frequent cases, it is located at the place of transition of the aortic arch into its descending section, i.e. quite far from the heart, next to the ductus arteriosus, on the inside of the back of the chest, to the left of the spine. There can be many variants of congenital narrowing of the aorta in this place, but in all of them there is an obstacle to blood flow, expressed to a greater or lesser extent, which determines the severity of the clinical manifestations of the defect.
With a very sharp narrowing of the aorta, the left ventricle, which pumps blood into the aorta, constantly works with increased load. As a result, blood pressure in the upper half (in the vessels of the neck, head, upper extremities) is significantly higher than in the lower half (in the vessels of the abdominal organs, lower extremities). The upper half of the body constantly lives with “hypertension,” and the lower half lives with constant hypotension. This does not mean that less blood enters the lower sections, no. Cardiac output remains sufficient. The heart copes, but due to an increase in the rhythm of contractions, an increase in the volume of the left ventricle, and thickening of its walls. These phenomena, especially with a sharp narrowing of the aorta, can lead to serious consequences.
In newborns and infants in the first months of life, coarctation of the aorta can be so severe that the blood supply to the lower sections is carried out only through the patent ductus arteriosus. The situation is very dangerous, critical. Signs of heart failure and hypertension in the upper half of the body, which threatens irreversible injury to the blood vessels of the brain, are increasing. This requires urgent intervention by a cardiac surgeon.
The operation itself consists of eliminating the area of narrowing of the aorta. This can be done in two ways: either completely remove the site of coarctation and then sew both healthy sections of the aorta together (“end to end”), or cut along the narrowed section and sew into it a patch from the child’s own subclavian artery or from synthetic material. Both methods are tried quite widely and, judging by the long-term results - 10-15 years or more - neither of them has any particular advantages. The fact is that in 20-25% of all patients operated on at an early age, narrowing may reappear at the operation site, regardless of the method. This may appear in a few years.
Today, thanks to the methods of X-ray surgery, it is possible to expand the re-narrowing by introducing a catheter with a balloon into the aorta. Pressure is created in the balloon and the narrowed area expands. This is often done during relapses. Also, a narrowed section of the aorta in older children can be stented, that is, a tubular structure, the so-called “stent”, is placed in the lumen of the aorta, which, with its walls as a frame, keeps the walls of the aorta from tending to narrowing and causing relapse of coarctation. But each of these methods has its own indications and contraindications, which your doctor will tell you about.
In any case, a child who has undergone surgery for coarctation in infancy should be under the supervision of cardiologists throughout his life. But at the same time, we must always remember that once, when he was very young, an operation saved his life.
In older children, even small ones, but not newborns, coarctation of the aorta can proceed quite calmly and be an accidental finding during the next medical examination (of course, if the pulse is felt not only in the arms, but also in the femoral arteries).
The indications for surgery will be not so much the diagnosis of coarctation itself, but rather circulatory disorders and the degree of arterial hypertension of the upper body.
The operation will be planned, and the choice of method depends on the specific situation. The operating surgeon will tell you about this. But you don't have to wait for many years. Coarctation will not go away and will not decrease, and the consequences of narrowing of the aorta will only increase. In adults, in advanced cases, surgery to eliminate coarctation is associated with a significantly greater risk of complications than in a preschool child or even a teenager.
Unfortunately, coarctation of the aorta cannot yet be eliminated by X-ray surgery (only its recurrence can be treated with this method). But technology is moving forward at an amazing speed and it may very well be possible that such treatment will become possible in the near future.
Symptoms
Symptoms depend on the severity of the disease. Symptoms in patients with significant narrowing appear already in childhood, while minor narrowing may not cause any changes in the body for quite a long time. Typically, severe cases manifest themselves immediately after birth. The child may suddenly become pale, irritable, sweating increases, and difficulty breathing occurs. If left uncorrected, the defect can lead to heart failure and subsequent death.
In older children and adults, the symptoms are erased, since the narrowing is insignificant. Often the only sign may be increased blood pressure measured in the arms. Other symptoms:
- Shortness of breath, mainly during physical activity;
- Headache;
- Muscle weakness;
- Coldness of the extremities;
- Nosebleeds;
Causes
Coarctation of the aorta is associated with disruption of the formation of the aorta during fetal development. Basically, the narrowing is located juxtaductally, which means next to the ductus arteriosus. The ductus arteriosus connects the aorta and the left pulmonary artery, and functions only in utero, closing after the onset of alveolar respiration. Probably, during the fetal period, part of the duct tissue moves to the aorta, involving its wall in the process of closure, which undoubtedly leads to narrowing.
Rarely, coarctation of the aorta occurs during life. Causes may include atherosclerotic lesions of the aorta, trauma, or Takayasu syndrome.
Risk factors
Patients with Shereshevsky-Turner syndrome have a genetic predisposition to coarctation. These people have only 45 chromosomes instead of 46, and in 1 out of 10 patients with this syndrome, coarctation of the aorta is detected.
Also, coarctation of the aorta can be combined with other congenital heart defects.
→Bivalve aortic valve;
→The aortic valve separates the aorta from the left ventricle. Normally it is tricuspid;
→Ventricular septal defect is a defect in which there is a communication between the right and left ventricles, as a result of which oxygenated blood from the left part mixes with venous blood from the right ventricle;
→Open ductus arteriosus. Normally, the ductus arteriosus closes immediately after birth; if it remains open, the defect is called patent ductus arteriosus.
→Stenosis of the opening of the aortic or mitral valves. A defect in which the openings of the mitral valve (the valve between the left atrium and the left ventricle) and the aortic valve (the valve between the left ventricle and the aorta) are reduced.
When to ask for help
If you have severe chest pain or sudden shortness of breath, you urgently need to contact your doctor or service 03; If you or your doctor have registered an unexplained increase in blood pressure that is difficult to control, it is necessary to be examined to determine if you have coarctation of the aorta, since timely treatment will avoid a dramatic development of events.
Diagnostics
The age at which pathology is detected depends on the severity of the disease. If the degree of narrowing is great, diagnosis is possible at an early age. Patients with minor narrowing often do not even know that they have an aortic defect. The disease is often an incidental finding.
A doctor may suspect a pathological narrowing of the aorta if:
- High blood pressure,
- Pressure differences in the arms and legs,
- Weak pulse in legs
- Heart murmur.
Instrumental research methods
Chest X-ray. There are certain radiological signs that allow you to establish the correct diagnosis).
Echocardiography. A method that allows you to visualize the defect, as well as recognize concomitant cardiac pathology, for example, bicuspid aortic valve
Electrocardiography. Since the heart muscle experiences a high load with this pathology, the doctor will use an electrocardiogram to determine signs of left ventricular hypertrophy.
Magnetic resonance imaging. High-resolution method to determine the exact location of coarctation.
Probing of the heart is also necessary to determine the severity of the lesion and the anatomical features of the defect.
Complications
If coarctation of the aorta is left untreated, you may experience complications over time:
- Stroke,
- Coronary artery diseases,
- Aortic rupture,
- Cerebral aneurysm,
Also, in addition, it should be said that the internal organs are poorly supplied with blood, since there is an obstacle to the flow of blood, which can gradually lead to degenerative changes in them with subsequent dysfunction.
Treatment of coarctation of the aorta
Surgery
There are several methods for resection and reconstruction of the aorta:
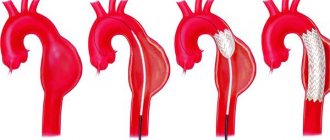
- Resection of aortic coarctation with end-to-end anastomosis. This method is possible in the presence of a narrowing of a short length, when it is possible to compare the ends of a normal, unchanged aorta.
- Aortic plastic surgery using a vascular prosthesis. This method is used if the narrowing covers a long segment and matching the ends is not possible. Instead of the resected pathological section of the aorta, a vascular prosthesis and special synthetic material are sewn in.
- Angiopalstastic using tissue from the left subclavian artery.
- Performing bypass surgery of the narrowed area. The shunt is sutured proximal and distal to the narrowed area, so that blood can freely bypass the pathological area of the aorta.
- Balloon angioplasty and stenting. This technique is often used if the area has narrowed after surgical correction. The doctor, using a guide, inserts a special balloon through the vessels into the aorta, which is inflated and the newly occurring narrowing is eliminated. Sometimes it is necessary to install stents (a kind of frame) that allow you to fix the required diameter of the vessel.
Drug treatment
Medications are not able to eliminate narrowing of the aorta, but are taken before surgical correction, and often for a long time after it, to control high blood pressure.
Postoperative period
In the postoperative period, it is possible that your blood pressure will remain elevated for some time and you will need to take antihypertensive drugs. Also, even several years after the operation, the reconstructed area (aneurysm) may expand or narrow again, which may require a repeat operation.
Prevention
If there is a high risk of this pathology in your family (there have been cases of this pathology in the family), you need to undergo medical and genetic counseling and determine with your doctor how high the risk of coarctation is in your children.
This is important to know!
During pregnancy, a woman with coarctation of the aorta should undergo careful monitoring by a cardiologist and obstetrician-gynecologist due to the possible development of complications from the cardiovascular system. And if there is an aneurysm of the reconstructed section of the aorta, then during pregnancy and childbirth this section may rupture. In this regard, strict monitoring of hemodynamic parameters, and especially your blood pressure, is necessary.
Patients with coarctation of the aorta are at risk for infective endocarditis, since altered aortic tissue in the area of coarctation is a favorable environment for the development of pathogenic organisms. Be sure to talk to your doctor about possible antibiotic prophylaxis before dental procedures to reduce the risk of endocarditis.
How to get treatment at the Scientific Center named after. A.N. Bakuleva?
Online consultations
Target. Due to the growing interest of clinicians in the correction of coarctation of the aorta from median sternotomy using cardiopulmonary bypass in newborns and infants, we retrospectively analyzed our own experience in resection and correction of coarctation with extended end-to-end anastomosis from a lateral approach with an emphasis on the frequency of reinterventions and the dynamics of growth of the transverse aortic arch in the long-term follow-up period.
Methods. The study included 124 patients under 3 months of age who underwent correction of aortic coarctation (from 2008 to 2021). In 43 (35%) patients, correction of coarctation was combined with a ventricular septal defect. 49 (39.5%) patients had hypoplasia of the transverse aortic arch (Z-score less than –2). All operations were performed using an extended end-to-end anastomosis via a lateral thoracotomy approach. In patients with a ventricular septal defect, narrowing of the pulmonary artery was undertaken simultaneously. The duration of postoperative follow-up was 3.6 (0.3–8.0) years.
Results. Hospital mortality was 1.6%. Survival rate during the long-term follow-up period is 93.5%. Aortic recoarctation was diagnosed in 10 (8%) patients after an average of 6.5 (3.5–15.0) months. after operation. Balloon angioplasty of the narrowing zone was effective in all cases and had no complications. We observed a statistically significant (p<0.001) increase in the transverse aortic arch in the group of patients with initial arch hypoplasia.
Conclusion. Resection for correction of coarctation with extended end-to-end anastomosis from a lateral approach is accompanied by low operative mortality, excellent survival and low reintervention rates. In a cohort of patients with initial moderate hypoplasia of the aortic arch, in the long-term follow-up period, growth of the arch to normal values was observed. Endovascular balloon dilatation of the area of re-narrowing of the aorta in the late postoperative period is a highly effective and safe procedure.
Introduction Surgical techniques for resection of aortic coarctation (CA) have changed significantly in the 70 years since this procedure was first described in 1945 [1, 2]. Initially, it consisted only of eliminating a narrowed section of the aorta. In connection with the development of neonatal cardiac surgery, there is a need to expand the capabilities of this technique to eliminate the entire range of problems with the aortic arch in newborns and infants, in particular with hypoplasia of the aortic arch. The first tangible successes along this path were achieved by teams that proposed the method of coronary artery resection with extended end-to-end anastomosis [3–5]. This method has been widely used in the world for 30 years due to its effectiveness and safety [3–8]. Somewhat later, attempts appeared to solve the problems of aortic arch pathology in infants using other surgical methods, in particular with the use of artificial circulation [9, 10]. The purpose of the study is to analyze our own experience in using the surgical technique of coronary artery resection with extended end-to-end anastomosis from a left-sided thoracotomy.
Methods Retrospective analysis was carried out on the medical records of all patients with coronary artery disease operated on in the Department of Cardiac Surgery and Intensive Care of the Children's City Clinical Hospital No. 13 named after. N.F. Filatova DZM" from July 14, 2008 to December 31, 2021. During this period, 231 patients were operated on using the method of coronary artery resection with extended end-to-end anastomosis via left posterolateral thoracotomy. Of these, 124 people who made up the present observation group were no more than 3 months old. The patient characteristics and anatomical features we assessed are presented below.
The study included patients with isolated coarctation of the aorta and coarctation in combination with ventricular septal defects or double-outlet right ventricle with subaortic ventricular septal defect. Patients in whom CA was combined with transposition of the great arteries, Taussig–Bing anomaly, and univentricular hemodynamics were excluded from the analysis. Twenty patients had a syndromic form of congenital heart disease, the data of which are presented below.
The main diagnostic method was transthoracic echocardiography. Absolute numerical values obtained from echocardiographic measurements were analyzed using the Boston Children's Hospital Z-score calculator (https://zscore.chboston.org). A Z-score less than –2 was used as a criterion for hypoplasia of the aortic arch and its parts [28]. There were 39% of such observations.
Surgical technique In all cases, coronary artery resection with extended end-to-end anastomosis was performed using an approach from the left posterolateral thoracotomy at the patient’s body temperature of 35 °C. A vascular clamp was applied to the aortic arch in such a way as not to disrupt the blood flow along the brachiocephalic trunk and to maintain a normal level of systolic blood pressure. An incision on the inner surface of the aortic arch was made up to the projection point of the mouth of the left common carotid artery. Anastomosis of the arch and the descending aorta was performed using a polypropylene suture with a 7/0 thread for 19 (15–33) minutes. When coarctation was combined with a large ventricular septal defect, during the same operation, the pulmonary artery narrowing procedure was performed using Mylar tape according to the Toronto-1 formula. There were 43 (35%) such observations.
Postoperative observation Data from the examination of patients in the postoperative period were obtained through a prospective survey or as a result of analysis of the database of follow-up observation of patients in the department. Routine examination included physical examination, weighing, height measurement, recording of blood pressure in the right arm and leg using the Korotkoff method, and transthoracic echocardiography. Transthoracic echocardiography protocols were available for analysis for 118 (95.2%) patients discharged from the hospital. Four children (3.2%) were not citizens of the Russian Federation, and after surgical treatment contact with them was lost. At the same time, all medical histories of children with aortic recoarctation, for which they underwent transluminal balloon angioplasty, as well as medical histories of children with concomitant ventricular septal defects, who underwent radical correction of the defect, were analyzed. Median postoperative follow-up was 3.6 (0.3–8.0) years.
Statistical analysis To sort variation series based on whether they correspond to a normal distribution, we used the one-sample Kolmogorov–Smirnov test. Normally distributed data are presented as means ± SD; not having a normal distribution - in the form of the median of a variation series indicating the minimum and maximum values. Comparison of average values was performed using the t-test, testing the hypothesis of equality of variances of the analyzed samples using the Livigne test. Survival and freedom from reoperation were analyzed using the Kaplan–Meier method. P<0.05 was accepted as statistically significant. Data were processed using SPSS Statistics 21.0 (IBM Corp., 2012). The ethical aspects of the study comply with the principles of the Declaration of Helsinki of the World Medical Association.
Results Immediate results In-hospital mortality in the observation group was 1.6% (n = 2). One of the patients, a premature baby weighing 1.54 kg, with a combination of coronary artery disease, multiple ventricular septal defects and moderate hypoplasia of the left ventricle with severe dysfunction, operated on for vital indications, died on the 2nd day. after intervention for acute heart failure due to the development of severe cardiac arrhythmias. In the second patient with trisomy 21, who died on the 5th postoperative day, CA was also combined with a ventricular septal defect, and the somatic condition was complicated by intrauterine infection and severe perinatal kidney damage. It should be noted that correction of isolated CA was accompanied by 100% survival. The average duration of mechanical ventilation after surgery was 26 hours, and the average duration of stay in the intensive care unit was 4 days. Not a single patient required unplanned re-interventions within 30 days. after resection of coarctation of the aorta. Transient complications of the early postoperative period were noted in 2.4% of cases: • neurological deficit with an episode of clonic-tonic seizures (n = 1; 0.8%); • left-sided chylothorax (n = 1; 0.8%), confirmed by cytological examination of the contents of the pleura; • paresis of the left recurrent laryngeal nerve (n = 1; 0.8%) with clinical stridor breathing, confirmed by fibrolaryngoscopy.
Results of a two-stage surgical strategy The median duration of the interstage observation period in 41 patients discharged from the clinic after removal of coronary artery disease and narrowing of the pulmonary artery was 9 (6–23) months. During this period, one child receiving tube feeding died at home as a result of aspiration of gastric contents. We noted spontaneous closure of the ventricular septal defect in 5 (11.6%) patients in this cohort; the second stage was to remove the band from the pulmonary trunk without cardiopulmonary bypass. Thirty-five patients underwent correction of the defect along with removal of the band under conventional cardiopulmonary bypass with superficial hypothermia. The duration of aortic cross-clamping and cardiopulmonary bypass in this group was 56 (34–117) and 106 (70–174) minutes, respectively. The median duration of mechanical ventilation after the second stage of correction was 22 (4–96) hours, stay in the intensive care unit after surgery was 2 (1–5) days. The second stage of correction was accompanied by 100% survival. Overall survival using a two-stage strategy in a group of 43 patients with a combination of coronary artery disease and septal defects was 93%.
Long-term results During 8 years of postoperative follow-up, survival rate in the observation group was 93.5% (Fig. 1).
Eight children died, four of whom had a syndromic form of congenital heart disease (Noonan, Holt-Oram, Down syndrome and 22q11.2 deletions), six had concomitant neurological and somatic diseases: cerebral palsy, epilepsy, hydrocephalic syndrome, chronic renal failure (after perinatal acute kidney injury), Hirschsprung's disease and duodenal atresia, corrected surgically. The death was associated either with the development of infectious complications during hospitalization in other hospitals in Moscow, or was the result of decompensation of concomitant diseases.
Freedom from interventions for aortic recoarctation over 8 years of postoperative follow-up was 92% (Fig. 2). In 10 (8%) patients, on average after 6.5 (3.5–15.0) months. After surgery, a narrowing of the aortic arch was diagnosed with an average systolic pressure gradient obtained by the Korotkov method of 42 (21–70) mm Hg. Art. Of these, 5 children were initially diagnosed with hypoplasia of the distal part of the transverse aortic arch with a Z-score of –4.0 (from –4.7 to –3.3).
All patients with repeated narrowing of the aorta underwent balloon angioplasty of the arch-descending aorta anastomosis zone. The mean pressure gradient recorded during aortic catheterization before the procedure was 37.0 ± 3.8 mmHg. Art.; after balloon dilatation - 8.2±1.4 mmHg. st., p = 0.03. No complications were noted. During a 3.5 (0.7–7.5) year follow-up in this cohort of patients, the average gradient between the arch and the descending aorta, recorded using transthoracic echocardiography, was 15.2 ± 3.3 mm Hg. Art. In the group of patients with initial hypoplasia of the aortic arch, we recorded a statistically significant increase in the diameter of the transverse arch 3.6 (0.3–8.0) years after the creation of an extended end-to-end anastomosis (Table 1).
Discussion
To date, several surgical techniques for correcting coronary artery disease have been described. This includes a simple end-to-end anastomosis [1, 2], Waldhausen isthmoplasty [12, 13], resection with reverse plastic surgery using a left subclavian artery flap [14], balloon dilatation of the narrowing zone [15, 16], and also a technique for correcting aortic coarctation under conditions of circulatory arrest or selective cerebral perfusion using access through a median sternotomy [9, 10]. We have been using the method of coronary artery resection with extended end-to-end anastomosis from a lateral approach for 10 years. In our study, we demonstrated that resection of aortic coarctation with extended end-to-end anastomosis using a lateral thoracotomy approach is accompanied by low operative mortality (1.6%), excellent long-term survival (93.5%) and low reintervention rate (8 %). A number of authors also prefer coronary artery resection from a lateral approach with an extended end-to-end anastomosis, considering the advantage of this method to be the possibility of complete resection of the entire coarctation zone with maximum excision of ductal tissue and the formation of a wide anastomosis of the arch and the descending aorta, which creates the opportunity for growth of the arch . They, as in this work, noted a low incidence of adverse outcomes and recoarctations (Table 2), which characterizes the safety and effectiveness of this method [6–8].
The European Congenital Heart Surgeons Association (ECHSA Congenital Database; echsacongenitaldb.org) contains information on the immediate results of 4,277 aortic coarctation resection operations with extended end-to-end anastomosis performed in European hospitals on newborns. and children of the first year of life with isolated CA and a combination of CA and ventricular septal defect from 1999 to the present. The graph was made using filters and database tools (Fig. 3). Given the fact that the department provides information on the operations performed on a regular basis to the ECHSA Congenital Database, we can state that the volume of interventions is above average with a mortality rate below average in the European hospitals participating in this project.
Up to 75% of patients requiring coronary artery removal have some degree of arch hypoplasia [3, 6, 7, 22]. In newborns, coarctation of the aorta is more often than in other age groups combined with hypoplasia of the arch of varying severity, which can be formed due to restriction of direct blood flow along the arch, including together with other intracardiac defects [23]. The growth potential of a hypoplastic arch has been sufficiently studied, including when removing coronary artery disease through resection and creating an extended anastomosis from a lateral approach [7, 24–26]. The authors demonstrated that even a hypoplastic arch adapts to the increased volume of blood flow, which is accompanied by its distinct growth [27, 28].
JY Liu and colleagues believe that it is necessary to evaluate the growth of the proximal and distal arches separately, since the distal arch shows more stable growth than the proximal arch [11]. The present study, however, demonstrated adequate growth of the hypoplastic aortic arch in both the proximal and distal transverse aortic arch. We believe that these data make it possible to achieve positive results in many observations to avoid operations with artificial circulation in the neonatal period, which can be accompanied by serious complications from the central nervous system that reduce the quality of life in general [29–32]. In patients with severe hypoplasia of the entire transverse aortic arch with a Z-score of less than –6, the question of the optimal method of surgical correction should be decided individually [33].
Among the complications of the presented method of surgical correction of coronary artery disease in the early postoperative period, bleeding, paresis of the recurrent nerve, sepsis, pulmonary hypertensive crisis, chylothorax, and neurological events in the form of convulsive syndrome are described [8]. We noted a small percentage (2.4%) of postoperative complications, which also positively characterizes this surgical technique. Aortic recoarctation is the most common complication of long-term follow-up after surgical correction of coronary artery disease. According to the literature, aortic recoarctation occurs in 4–19% of patients after operations during infancy [11, 17, 21]. The reasons for the development of this complication are not fully understood. Researchers believe that the more frequent occurrence of re-narrowing of the aortic lumen may be associated with a high initial systolic blood pressure gradient, defects in surgical technique, the presence of residual ductal tissue in the aortic lumen, or neointimal proliferation [11, 21, 34]. The occurrence of repeated narrowing can also, of course, be influenced by the initial anatomy with arch hypoplasia, the degree of its severity and extent, and options for surgical correction techniques [6, 34]. The highest incidence of aortic recoarctation is observed in children operated on before the age of 1 year, regardless of the type of surgical technique [35, 36]. Analyzing the frequency of re-narrowing of the aorta after coronary artery resection from a lateral approach, we noted a low incidence of this complication, which is comparable with the data of other researchers [18–21, 33] (Table 2). When analyzing balloon angioplasty of the area of re-narrowing of the aorta in the long-term postoperative period, we, like other researchers, noted high efficiency and technical simplicity in eliminating aortic recoarctation [20, 37]. In patients with coarctation and ventricular septal defect, the two-stage surgical strategy was also associated with excellent clinical results in this patient population. This strategy allowed us to avoid surgery with cardiopulmonary bypass in the neonatal period and enabled patients with concomitant perinatal, neurological and somatic pathology between the two surgical interventions to achieve sustainable compensation, which, there is reason to believe, improved the results of surgical treatment.
Limitations The study is limited by its retrospective nature and the sample of patients studied. Characteristics of the results of coronary artery resection with extended end-to-end anastomosis are given in the mid-term follow-up period, while the anatomical characteristics were analyzed using only transthoracic echocardiography. Information obtained using angiography and magnetic resonance imaging in this group of patients may contribute to further detailed study of the results of the method. The study of long-term results of CA removal using the resection method with extended end-to-end anastomosis is a prospective process; we will continue their analysis, including with an emphasis on the potential risk of arterial hypertension, especially in the group of children with initial hypoplasia of the transverse arch and patients with repeated narrowing of the aorta.
Conclusion: CA resection with extended end-to-end anastomosis from a lateral approach is associated with low operative mortality, excellent survival, and a low rate of reintervention during 8 years of postoperative follow-up. In a cohort of patients with initial hypoplasia of the transverse aortic arch, growth of the arch to normal values was noted. Endovascular balloon dilatation of the area of re-narrowing of the aorta in the late postoperative period is an effective and safe procedure.
Funding The study had no sponsorship.
Conflict of interest The authors declare no conflict of interest.
Authors' contributions Concept and design: V.N. Ilyin Data collection and analysis: M.M. Belyaeva, O.Yu. Kornoukhov, Yu.Yu. Kornoukhov, O.I. Kalinina Article writing: M.M. Belyaeva Correction of the article: V.N. Ilyin., O.Yu. Kornoukhov Approval of the final version for publication: M.M. Belyaeva, V.N. Ilyin, O.Yu. Kornoukhov, Yu.Yu. Kornoukhov, O.I. Kalinina
CoA
Coarctation of the aorta is a vascular malformation consisting of narrowing or complete closure of the lumen of the aorta in a limited area.
In this case, a violation of general blood circulation occurs.
Coarctation of the aorta is one of the most common congenital heart defects.
Taking into account the localization of the pathological narrowing, coarctation is distinguished in the area of the isthmus, ascending, descending, thoracic, and abdominal aorta. Some sources identify the following anatomical variants of the defect: preductal stenosis (narrowing of the aorta proximal to the confluence of the PDA) and postductal stenosis (narrowing of the aorta distal to the confluence of the PDA).
According to the criterion of multiplicity of anomalies of the heart and blood vessels, A.V. Pokrovsky classifies 3 types of aortic coarctation:
type 1 - isolated coarctation of the aorta (73%);
type 2 – combination of aortic coarctation with PDA; with arterial or venous blood discharge (5%);
Type 3 – a combination of aortic coarctation with other hemodynamically significant vascular anomalies and congenital heart disease (12%).
In the natural course of coarctation of the aorta, 5 periods are distinguished:
I (critical period) - in children under 1 year; characterized by symptoms of circulatory failure in the pulmonary circulation; high mortality from severe cardiopulmonary and renal failure, especially when aortic coarctation is combined with other congenital heart defects.
II (adaptive period) - in children from 1 to 5 years; characterized by a decrease in the symptoms of circulatory failure, which is usually represented by increased fatigue and shortness of breath.
III (compensatory period) – in children from 5 to 15 years; characterized by a predominantly asymptomatic course.
IV (period of development of relative decompensation) – in patients 15-20 years old; During puberty, signs of circulatory failure increase.
V (period of decompensation) – in patients 20-40 years old; characterized by signs of arterial hypertension, severe left- and right-ventricular heart failure, and high mortality.
Prognosis of aortic coarctation
The natural course of aortic coarctation is determined by the type of aortic narrowing, the presence of other congenital heart defects, and in general has an extremely unfavorable prognosis. In the absence of cardiac surgery, 40-55% of patients die in the first year of life. With timely surgical treatment of aortic coarctation, good long-term results can be achieved in 80-95% of patients, especially if the operation was performed before the age of 10 years.