SYSTEMIC AMYLOIDOSIS: diagnosis, differential diagnosis, treatment
“Amyloidosis” is a term that unites a group of diseases that are distinguished by a wide variety of clinical manifestations and are characterized by extracellular deposition of insoluble pathological fibrillar proteins in organs and tissues. This pathology was first described in the 17th century. Bone - sago spleen in a patient with liver abscess. In the middle of the 19th century. Virchow used the botanical term "amyloid" (from the Greek amylon - starch) to describe the extracellular material found in the liver at autopsy, since he believed that it was similar in structure to starch. Subsequently, the protein nature of the deposits was established, but the term “amyloid” has been preserved to this day.
In the 20s In the 20th century, Benhold proposed staining amyloid with Congo red, then the effect of birefringence in polarized light was discovered - a change in brick-red color to apple-green. In 1959, Cohen and Calkins used electron microscopy to establish the fibrillar structure of amyloid.
Clinical ideas about amyloidosis also underwent evolution: Rokitansky in 1842 established a connection between “sebaceous disease” and tuberculosis, syphilis, and rickettsiosis; Wilkes in 1856 described “fat organs” in a patient who had no concomitant diseases; Atkinson discovered amyloidosis in patients with multiple myeloma in 1937. Senile (Soyka, 1876) and hereditary (Andrade, 1952) forms of the disease were identified, amyloidosis was divided into genetic, primary and secondary, and finally, in 1993, the WHO classification was adopted, based on the specificity of the main fibrillar protein of amyloid.
In our country, E. M. Tareev, I. E. Tareeva, V. V. Serov made a great contribution to the development of ideas about amyloidosis. A huge role in the study of primary and genetic variants of amyloidosis and periodic disease belongs to O. M. Vinogradova, whose monographs, published in 1973 and 1980, have not lost their relevance today.
Currently, amyloidosis is clinically divided into systemic and local forms. Among the systemic forms, depending on the composition of fibrillar deposits, four types are distinguished (Table 1).
Local forms of amyloidosis currently include Alzheimer's disease (A-beta, fibrils consist of β-protein deposited in the brain), amyloidosis of the pancreatic islets, possibly having a pathogenetic connection with type 2 diabetes, amyloidosis arising in endocrine tumors, amyloid tumors of the skin, nasopharyngeal region, bladder and other rare types.
AL amyloidosis
The development of AL amyloidosis is possible in myeloma, Waldenström's disease, B-cell lymphomas, and it can be idiopathic in primary amyloidosis. All these options are united by a common pathogenesis; primary amyloidosis is the most difficult to recognize due to the absence of obvious signs of a hematological disease, so it is this form that is worth dwelling on in detail.
In primary amyloidosis, a benign plasma cell dyscrasia related to multiple myeloma, abnormal clones of bone marrow plasma cells produce amyloidogenic immunoglobulins. Some amino acids in the variable regions of the light chains of these immunoglobulins occupy an unusual position, which leads to their instability and causes a tendency to fibrillogenesis. In patients with primary amyloidosis, the content of plasma cells in the bone marrow is increased to 5-10% (normally less than 4%, in myeloma - more than 12%), and they produce a certain isotype of immunoglobulin light chains, which predominates in immunohistochemical staining. Free monoclonal light chains of the predominant lambda or (less commonly) kappa isotype are detected in the blood and urine, but their content is lower than in multiple myeloma.
The clinical picture of primary amyloidosis is diverse and is determined by the predominant involvement of certain organs in the pathological process - the heart, kidneys, nervous system, gastrointestinal tract, liver, etc. The first symptoms are weakness and weight loss, but at this stage, before the appearance of organ symptoms , the diagnosis is made extremely rarely.
The target organs for AL amyloidosis most often are the kidneys and heart. Kidney damage is manifested by nephrotic syndrome, which persists even with the onset of chronic renal failure; hematuria and arterial hypertension are not typical.
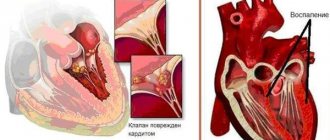
When amyloid is deposited in the myocardium, various rhythm disturbances and progressive heart failure develop, which may be preceded by asymptomatic changes on the ECG in the form of a decrease in wave voltage. Echocardiographic examination reveals concentric thickening of the walls of the left and right ventricles, a decrease in the volume of the heart cavities, a moderate decrease in ejection fraction, and diastolic dysfunction of the left ventricular myocardium.
Symptoms of nervous system involvement are often observed - autonomic, in the form of orthostatic hypotension, and peripheral, in the form of sensitivity disorders. In recent years, lesions of the central nervous system have also begun to be described, although previously it was believed that they were not characteristic of primary amyloidosis.
Dyspeptic symptoms (feeling of fullness, constipation, diarrhea) and malabsorption syndrome can be caused by both damage to the autonomic nervous system and amyloidosis of the gastrointestinal tract. Hepatomegaly is very characteristic, the nature of which should be differentiated between congestion due to heart failure and amyloid liver damage. The latter is confirmed by an increase in serum alkaline phosphatase levels. The spleen is often affected, but splenomegaly is not always detected and is not of great clinical significance.
Macroglossia, a classic sign of primary amyloidosis, is observed in 20% of patients; infiltration of soft tissues can lead to atrophy of muscles, skin, nail dystrophy, alopecia and the appearance of tumor-like formations - amyloid.
Less common is vascular lesions, the symptoms of which are periorbital purpura - “raccoon eyes” and ecchymoses. Bleeding may occur, including bladder bleeding, caused by both changes in the vascular wall and a violation of the coagulation system, primarily a deficiency of the X factor, which binds to amyloid. The thrombocytosis characteristic of amyloidosis is also commonly explained by a deficiency of coagulation factors.
Pulmonary amyloidosis is often discovered only at autopsy. However, in some cases, shortness of breath, hemoptysis and hydrothorax may be caused not only by congestive heart failure and nephrotic syndrome, but also by amyloid lung disease. Deposition of amyloid in the alveoli and the development of pulmonary amyloid are possible. X-rays can reveal reticular and nodular changes in the lung tissue.
Damage to the adrenal glands can lead to adrenal insufficiency, which often remains unrecognized, since hypotension and hyponatremia are considered symptoms of heart failure and damage to the autonomic nervous system. In 10-20% of patients, hypothyroidism may occur as a manifestation of damage to the thyroid gland; enlargement of the submandibular salivary glands is often encountered.
The diagnosis of primary amyloidosis, in addition to the indicated clinical features, which may be similar in secondary amyloidosis, is based on a number of laboratory data. In 85% of patients, immunoelectrophoresis of serum and urine proteins reveals monoclonal immunoglobulins. In routine studies, the same monoclonal immunoglobulins are detected in the urine in the form of Bence Jones protein. Bone marrow biopsy allows for a differential diagnosis of multiple myeloma and also reveals a moderate increase in the number of plasma cells and their monoclonality with immunohistochemical staining.
However, even the combination of a characteristic clinical picture and the presence of monoclonal plasma cells and proteins is not yet sufficient to confirm the diagnosis of primary amyloidosis. Biopsy data play a decisive role here. The least invasive is aspiration of the subcutaneous fatty tissue of the anterior abdominal wall, which gives 80-90% positive results in AL amyloidosis (this method has not yet found application in our country). A biopsy of the gums and rectal mucosa has a certain diagnostic value, but the percentage of positive results varies widely, depending on the stage of the process, so it is advisable to perform a biopsy of one of the affected organs - the kidney, liver, heart, which gives almost 100% positive results for amyloidosis AL-type.
First of all, the biopsy material is stained with Congo red. If congophilia of the material under study is detected, it must be examined in polarized light; the effect of birefringence is characteristic only of amyloid; other congophilic substances do not acquire an apple-green color. After this, amyloid typing is desirable. The most accurate is the immunohistochemical method using monoclonal antibodies to amyloid precursor proteins. However, at present it is practically unavailable in our country. Therefore, for diagnosis, staining with solutions of alkaline guanidine or potassium permanganate is used, which allows, although indirectly, to determine the type of fibrillar deposits.
The prognosis for primary amyloidosis is worse than for other forms of the disease, the average life expectancy does not exceed two years, in the presence of heart damage or multisystem damage without treatment, patients die within a few months. The most common causes of death are cardiac and renal failure, sepsis, vascular complications and cachexia. Pathogenetic similarity with multiple myeloma allows one to expect inhibition of disease progression with chemotherapy administered to suppress monoclonal plasma cells. There are several treatment regimens (Table 2).
The use of chemotherapy, if treatment is successful, can increase the life expectancy of patients by 10 to 18 months. But the effectiveness of therapy is low, in particular due to the fact that in many cases the progression of the disease leads to the death of patients before completing the course of treatment, as well as due to the development of cytopenia, infectious complications, and fatal rhythm disturbances during treatment with ultra-high doses of dexazone. The use of high doses of melfolan with autologous stem cell transplantation allows achieving remission in more than 50% of cases, however, the use of this method is limited by the severity of the condition, the age of the patients, and functional disorders of the heart and kidneys. In many cases, only symptomatic maintenance therapy is possible.
AA amyloidosis
The development of AA amyloidosis occurs during chronic inflammatory processes; the precursors of AA amyloid are serum acute phase proteins, α-globulins, produced by different types of cells, mainly neutrophils and fibroblasts. Secondary amyloidosis develops with rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, various tumors, lymphogranulomatosis, ulcerative colitis and Crohn's disease, with periodic disease (familial Mediterranean fever), as well as with tuberculosis, osteomyelitis, bronchiectasis.
Characteristic clinical features of AA amyloidosis are kidney damage in most patients, as well as relatively rare damage to the liver and/or spleen (about 10%) and the heart (detected only by echocardiography). Macroglossia is not typical for secondary amyloidosis. The diagnosis is based on a combination of renal amyloidosis and chronic inflammatory disease, confirmed by immunohistochemical staining of biopsy material; in our country, the indirect staining methods already mentioned above are used.
The prognosis largely depends on the nature of the underlying disease; during the natural course, a third of patients develop renal failure 5 years from the moment proteinuria is detected. For periodic disease, the five-year survival rate is 25%.
Treatment is based on suppression of the focus - the source of production of serum precursor proteins. Removal of tumors, sequestrectomy, bowel resection, treatment of tuberculosis, reduction of the activity of rheumatoid arthritis (with the use of cytostatics) lead to the cessation of the progression of amyloidosis, and sometimes to the reverse development of clinical manifestations, in particular nephrotic syndrome.
The use of colchicine for periodic illness is the method of choice, its effectiveness has been proven, treatment prevents the development of amyloidosis and inhibits its progression. In other forms of secondary amyloidosis, the effectiveness of colchicine has not been confirmed.
Senile and hereditary forms of systemic amyloidosis, as well as local forms, are rare; dialysis amyloidosis is well known to specialists and is practically never encountered in general practice.
Symptomatic therapy depends not on the type of amyloidosis, but on the affected target organs (Table 3).
Amyloidosis, especially primary, is considered an uncommon pathology, but in reality it is not so rare as it is difficult to diagnose. Adequate diagnosis requires not only knowledge of the clinical picture and pathogenesis of the disease, but also the availability of certain diagnostic capabilities. To illustrate this point, we present our own data (see Table 4). In the nephrology department of the S.P. Botkin Moscow City Clinical Hospital in 1993-2003. 88 patients were observed who were diagnosed with amyloidosis.
The diagnosis was confirmed morphologically in all patients with AL amyloidosis, senile and unspecified amyloidosis, and in 30 patients with secondary amyloidosis - in a total of 53 cases. In 12 patients a kidney biopsy was performed, in two patients a liver biopsy was performed, in eight patients a bowel biopsy was performed, in 12 cases a gum biopsy was performed, and in another 19 cases the diagnosis was confirmed by morphological examination of sectional material.
In most cases, the diagnosis of amyloidosis was made for the first time as a result of examination in the nephrology department. We made a comparison among patients with AL amyloidosis of referral and clinical diagnoses (Table 5).
In only two cases out of 20 (10%) the referral diagnosis was “primary amyloidosis”, and one of these patients was diagnosed at the MMA therapy and occupational diseases clinic, and the other at a foreign clinic.
All patients who were diagnosed with myeloma with the development of AL amyloidosis were transferred to hematology departments. Of the 11 patients with primary amyloidosis, seven patients received chemotherapy with a combination of melfolan and oral prednisolone in intermittent courses, four of them in combination with dialysis treatment, and another patient received only dialysis and symptomatic treatment. Of these patients, five people died within a period of two weeks to two years from the start of treatment (all with renal failure and multiple organ damage), one patient is on dialysis, one patient was referred for autologous stem cell transplantation, and one patient is receiving treatment until present time. In one patient, chemotherapy was postponed due to the presence of a long-lasting gastric ulcer, and two other patients refused treatment.
Among patients with secondary amyloidosis in our study, patients with rheumatoid arthritis predominated; chronic osteomyelitis and psoriatic arthritis were in second place among the causes; other diseases were less common (Table 6).
Treatment of rheumatoid arthritis and psoriatic arthritis was carried out with the use of cytostatics (metatrexate, azathioprine), although in many cases the treatment options were limited due to the presence of chronic renal failure and concomitant pathology. Patients with chronic osteomyelitis were sent to purulent surgery departments. Patients with ankylosing spondylitis and Crohn's disease received specific treatment, patients with COPD and tuberculosis were also sent to specialized hospitals. One of the patients with a stomach tumor was successfully operated on, and over the course of four years of observation, the nephrotic syndrome gradually regressed; in other cases of tumors, the prevalence of the process allowed only symptomatic therapy; the patient with lymphogranulomatosis was admitted in a terminal condition. The mortality rate among patients with secondary amyloidosis was 38% (due to patients with advanced lesions at the time of diagnosis). All patients with periodic illness received colchicine therapy.
Features of the diagnosis and application of modern methods of treatment of primary amyloidosis can be illustrated by the following example: patient K., 46 years old, was first hospitalized at the end of October 2002 with complaints of swelling in the legs, palpitations, and amenorrhea. The history includes colds, an appendectomy, two normal emergency deliveries, no indications of kidney disease, and no chronic diseases. In April 2002, she suffered acute pneumonia in the upper lobe of the right lung, was treated as an outpatient, and received injections of abactal and lincomycin. Due to the localization of pneumonia, she was examined at a tuberculosis clinic, and the diagnosis of tuberculosis was excluded. At the beginning of June, swelling in the legs first appeared, for which she was not examined. The swelling disappeared on its own after a short time and then resumed. The patient was hospitalized in a therapeutic hospital; examination revealed proteinuria up to 1.65%, hypoproteinemia (total serum protein 52 g/l), normal blood pressure (120/80 mm Hg), unchanged urinary sediment, creatinine plasma levels are also within normal limits. A diagnosis of acute glomerulonephritis was made, treatment with ampicillin, chimes, heparin, triampur was performed, and a tonsillectomy was performed. Proteinuria persisted, edema gradually increased, and therefore, for further examination and treatment, the patient with a diagnosis of chronic glomerulonephritis was sent to the hospital named after. S. P. Botkina.
On examination, the skin is clean, of normal color, anasarca, massive, dense edema, ascites is detected, peripheral lymph nodes are not enlarged. Blood pressure 110/70 mm Hg. Art., heart sounds are sonorous, clear, rhythmic, heart rate 90 beats/min, liver and spleen are not enlarged, diuresis is up to 1000 ml/day, stool is regular, without pathological impurities. The examination revealed nephrotic syndrome - proteinuria 3 g/l, scanty urinary sediment, hypodysproteinemia, hyperlipidemia (total serum protein 39 g/l, albumin 12 g/l, globulins 7-30-15-19%, respectively α1-α2-β -γ cholesterol 17.8 mmol/l, β-lipoproteins 250 units), when analyzing urine for Bence-Jones protein - the reaction is negative, daily excretion of 17-KS is not reduced. Clinical blood test and other biochemical parameters are within normal limits, coagulogram shows severe hyperfibrinogenemia, increased level of RKFM. Study of blood immunoglobulins: Ig-A - 0.35, Ig-M - 35.7 (two norms), Ig-G - 1.96 g/l. X-ray of the chest organs, skull and pelvic bones, ultrasound of the abdominal cavity, kidneys, thyroid gland, ECHO-CG without pathology, pelvic ultrasound - signs of adenomyosis of the uterine body, endoscopy - reflux esophagitis, chronic gastritis. When examined by a neurologist, no pathology was found; the oncologist diagnosed fibrocystic mastopathy.
In order to clarify the genesis of nephrotic syndrome, a fine-needle puncture biopsy of the right kidney was performed under local anesthesia and ultrasound guidance; there were no complications. When examining a biopsy specimen, amyloid deposition is noted in the glomerular mesangium and in extraglomerular vessels. Amyloid loads up to 25% of glomerular vascular loops. An immunohistochemical study did not find any specific luminescence. When the preparations are treated with an alkaline guanidine solution for 2 hours, congophilia and their properties in polarized light are preserved, which is characteristic of AL amyloidosis.
To clarify the nature of AL amyloidosis, an immunochemical study of blood and urine was performed in the Immunotest laboratory. M-lambda paraproteinemia with a decrease in the level of polyclonal immunoglobulins and Bence-Jones lambda-type paraproteinuria against the background of massive non-selective proteinuria were detected. The patient was consulted by a hematologist, the presence of Waldenström's disease was suggested, and a bone marrow biopsy was performed. Conclusion: in the existing bone marrow cavities, cells of all three germs of normal hematopoiesis are visible, as well as lymphoid cells that do not form clusters. The diagnosis of Waldenström's disease was rejected due to the absence of lymphoid infiltration of the bone marrow, enlargement of the lymph nodes and spleen, and the absence of tumor substrate.
A diagnosis of primary amyloidosis with kidney damage, nephrotic syndrome, preserved renal function was established; no signs of other organ damage were detected. In January 2003, chemotherapy was started with melfolan 16 mg/day and prednisolone 100 mg/day, courses of four days every six weeks. Symptomatic treatment is also carried out: furosemide, veroshpiron, potassium preparations, famotidine, albumin transfusions. To date, five courses of chemotherapy have been carried out with good tolerance, edema has decreased, proteinuria has decreased to 1.8 g/l, and the severity of hypodysproteinemia has slightly decreased (total protein 46 g/l, albumin 18 g/l, α2-globulins 20%). Kidney function remains intact, plasma creatinine is 1.3 mg/dL, and no signs of damage to other organs and systems were detected during dynamic control examinations.
This case clearly illustrates the fact that morphological, immunological and immunochemical examination is necessary to diagnose amyloidosis. Thus, in our patient, the most obvious clinical diagnosis was “chronic glomerulonephritis,” and in the absence of the possibility of performing a kidney biopsy, this diagnosis would most likely have been made. The patient did not have any clinical indications of the systemic nature of the disease, a chronic inflammatory process, or a disease of the blood system, with the exception of an increase in the level of Ig-M. And only the data obtained from the study of the renal biopsy entailed a bone marrow trephine biopsy and an immunochemical study, which together made it possible to make a diagnosis of primary amyloidosis before the appearance of systemic damage. Pathogenetic therapy was started, although against the background of already developed nephrotic syndrome, but before the onset of renal failure and when only 25% of the glomeruli were loaded with amyloid, which has a relatively favorable prognosis.
In conclusion, we note that amyloidosis is a serious disease with a high mortality rate, which is extremely difficult to diagnose, however, timely and high-quality examination of patients makes it possible to make a diagnosis earlier, and the timely prescription of adequate therapy, in turn, makes it possible to improve the prognosis in this case. group of patients.
Literature
- Varshavsky V. A., Proskurneva E. P. Significance and methods of morphological diagnosis of amyloidosis in modern medicine // Practical Nephrology. - 1998. - 2:16-23.
- Vinogradova O. M. Primary and genetic variants of amyloidosis. - M.: Medicine, 1980.
- Zakharova E. V., Khrykina A. V., Proskurneva E. P., Varshavsky V. A. A case of primary amyloidosis: difficulties in diagnosis and treatment // Nephrology and Dialysis. - 2002. - 1:54-61.
- Rameev V.V. Features of kidney damage in AA and AL amyloidosis: dissertation ... cand. honey. Sci. - M., 2003.
- Kozlovskaya L.V., Varshavsky V.A., Chegaeva T.V. et al. Amyloidosis: a modern view of the problem // Practical Nephrology. - 1998. - 2:24-26.
- Rodney H., Raymond LC and Skinner M. // The systemic Amyloidoses; New England Journal of Medicine, 1997. - 337:898-909.
- Dhodapkar M.V., Jagannath S., Vesole d. et al // Treatment of AL-amyloidosis with dexamethasone plus alpha interferon / Leuc Lymphoma. 1997. - 27(3-4):351-365
- Gertz MA, Lacy MQ, Lust JA et all // Phase II trial of high-dose dexamethasone for previosly treated immunoglobulin light-chain amyloidosis. Am J Hematol 1999,61(2):115-119.
- Gertz MA, Lacy M., Q., Lust JA et al // Phase II trial of high-dose dexamethasone of untreated patients with primary systenic amyloidosis. Med Oncol 1999.- 16(2):104-109
- Sezer O., Schmid P., Shweigert M. et al // Rapid reversal of nephrotic syndrome due to primary systemic AL amyloidosis after VAD and subsequent high-dose chemotherapy with autologous stem sell support. Bone Marrow Transplant. 1999. - 23(9): 967-969.
- Sezer O., Neimoller K., Jakob C. et al // Novel approaches to the treatment of primary amyloidosis. Expert Opin Investig Grugs. 2000. - 9(10):2343-2350
- Sezer O., Eucker J., Jakob C., Possinger K. // Diagnosis and treatment of AL amyloidosis. Clin Nephrol. 2000. - 53(6):417-423.
- Skinner M. "Amyloidosis" Current Therapy in Allergy, Immunology, and Rheumatology. Mosby-Year Book. 1996. - 235-240.
- Palladini G., Anesi E., Perfetti V. et al. A modified high-dose dexamethasone regimen for primary systemic (AL) amyloidosis. British Journal of Haematology. 2001. - 113:1044-1046.
E. V. Zakharova Moscow City Clinical Hospital named after. S. P. Botkina
Table 2. Treatment regimens for primary amyloidosis
- Cyclic oral administration of melfolan (0.15-0.25 mg/kg body weight per day) and prednisolone (1.5-2.0 mg/kg per day) for four to seven days every four to six weeks for a year, up to achieving the course dose of 600 mg
- Oral administration of melfolan at a dose of 4 mg/day for three weeks, then, after a two-week break - 2-4 mg/day four days a week continuously, until a course dose of 600 mg is reached, in combination with prednisolone
- Intravenous administration of high doses of melfolan (100–200 mg/m² body surface for two days) followed by autologous stem cell transplantation
- Intravenous dexamethasone 40 mg for four days every three weeks—eight cycles
- Intravenous administration of dexamethasone at a dose of 40 mg on the first-fourth, 9-12th and 17-20th days of a 35-day cycle, three to six cycles, followed by the use of a-interferon at a dose of 3-6 million units three times a day a week
- Vincristine-doxoribucin-dexamethasone (VAD) regimen
Cardiac dysfunction
Intracardiac hemodynamic disturbances in AS are caused mainly by diastolic dysfunction; the decrease in myocardial contractility is usually insignificant. In a study that included 223 patients with AS, the mean LV ejection fraction was 52.5 ± 13.1% in patients with AL amyloidosis and 58 ± 13% in patients with hereditary ATTR amyloidosis. It was significantly lower only among patients with senile ATTR amyloidosis: 44.2 ± 15.4% [16]. A decrease in ejection fraction of less than 40% was noted in 40% of patients with senile ATTR amyloidosis and only in 22% of patients with AL amyloidosis and 8% of patients with hereditary ATTR amyloidosis. Other researchers have described similar changes in contractile function [18].
Diastolic dysfunction of varying degrees is detected in almost all patients. Early in the course of the disease, amyloid deposits impair isovolumic relaxation of the myocardium, resulting in a decrease in the rate of early diastolic LV filling (E) and an increase in late diastolic flow (A). Thus, a decrease in the E/A ratio (type 1 diastolic dysfunction) is an early sign of heart damage. As AS progresses, the myocardial wall becomes stiffer, the pressure in the left atrium increases, which leads to an increase in the rate of early diastolic filling and, thus, to a pseudo-normalization of the E/A ratio (type 2 diastolic dysfunction) [2]. With a further increase in the stiffness of the heart wall and an increase in end-diastolic pressure, a significant decrease in late diastolic flow occurs, which makes it possible to identify a decrease in myocardial compliance (3rd restrictive type of diastolic dysfunction). Restrictive changes are often accompanied by dilatation of both atria [3, 18].
Although restrictive disorders of diastolic function are considered the most typical for AS, research data show the possibility of developing other disorders of intracardiac hemodynamics [34–36].
Low salt diet
A low-salt diet is associated with slower spread and decreased severity of amyloidosis throughout the body. When amyloid plaques are deposited in the body, it is important to eat a healthy diet.
Get more rest
With amyloidosis, organ systems have difficulty performing normal functions. Heavy stress can aggravate the condition and cause faster organ breakdown and worsen tissue function. You will probably have to change your lifestyle. This way, amyloidosis can be slowed down, which can literally add years to your life.
A high fiber diet improves health
The digestive tract is one of the main targets for the deposition of abnormal proteins. Therefore, the gastrointestinal tract of patients with amyloidosis is especially fragile and prone to damage. A high-fiber diet improves bowel function and also improves the absorption of nutrients from the foods you eat. To improve your gut health, it is important to eat a fiber-rich diet.
Diagnostics
Taking into account the fact that amyloidosis can affect various organs, and the clinical picture of the disease is diverse, its diagnosis presents certain difficulties. The functional state of internal organs can be assessed by:
- Ultrasound;
- EchoCG;
- ECG;
- radiography;
- gastroscopy (EGD);
- sigmoidoscopy.
The incidence of amyloidosis is 1 case per 50,000 people. The disease is more common in older people.
Amyloidosis can be suspected if the following changes are detected in laboratory test results:
- anemia;
- thrombocytopenia;
- hypocalcemia;
- hyponatremia;
- hyperlipidemia;
- hypoproteinemia;
- cylindruria;
- leukocyturia.
For final diagnosis, it is necessary to perform a puncture biopsy of the affected tissues (mucosa of the rectum, stomach, lymph nodes; gums; kidneys) followed by histological examination of the obtained material. The detection of amyloid fibrils in the test sample will confirm the diagnosis.
Forecast
Amyloidosis is a chronic, progressive disease. In secondary amyloidosis, the prognosis is largely determined by the possibility of treating the underlying disease.
As complications develop, the prognosis worsens. Once symptoms of heart failure appear, the average life expectancy is usually less than a few months. The life expectancy of patients with chronic renal failure is on average 12 months. This period increases slightly in the case of hemodialysis.
Forms of the disease
Depending on the reasons that caused it, amyloidosis is divided into several clinical forms:
- senile (senile);
- hereditary (genetic, family);
- secondary (acquired, reactive);
- idiopathic (primary).
Depending on which organ amyloid deposits are predominantly deposited, the following are distinguished:
- amyloidosis of the kidneys (nephrotic form);
- cardiac amyloidosis (cardiopathic form);
- amyloidosis of the nervous system (neuropathic form);
- liver amyloidosis (hepapathic form);
- adrenal amyloidosis (epinephropathic form);
- ARUD-amyloidosis (amyloidosis of organs of the neuroendocrine system);
- mixed amyloidosis.
Kidney amyloidosis
Amyloidosis can also be local or systemic. With local amyloidosis, there is a predominant lesion of one organ, with systemic amyloidosis - two or more.
Procedures and operations
Transplantation of a donor organ is the only possible way to save the life of a patient with organ failure that has developed against the background of amyloidosis. It is important to understand that transplantation is only a symptomatic treatment method that is not able to eliminate the true cause and slow down the progression of amyloidosis. Even after transplantation, recurrence of the disease is possible.
What can be transplanted for amyloidosis:
- heart;
- kidney;
- skin;
- liver tissue.
A donor organ can be obtained either from a living donor or from a person who has been diagnosed with brain death, but at the same time retains the functional activity of the internal organs. To date, an artificial heart has been developed, which is presented in the form of a fully mechanized device that pumps blood in the human body. After transplantation, lifelong use of immunosuppressants is prescribed, which helps prevent rejection of the “foreign” tissue or organ.
Effective folk remedies for protection against amyloidosis
Red wine component resveratrol protects against amyloidosis
Scientists have found that the red wine component resveratrol has some preventive measures against the development and severity of amyloidosis. While resveratrol is considered an allergen for some individuals, it also has many positive health benefits. Drinking 1-2 glasses of red wine weekly is probably enough to prevent amyloidosis.
Plants containing germanium are effective remedies for amyloidosis
Some plants contain the microelement germanium, which has a positive preventive effect on amyloidosis and its consequences throughout the body. These folk remedies are promising alternative treatments for amyloidosis.
During pregnancy
Pregnancy with amyloidosis is allowed only if the functional activity of the woman’s vital organs allows her to bear and give birth to a healthy child. Otherwise, pregnancy can cause death not only for the fetus, but also for the mother herself.
Certain local forms of amyloidosis do not prevent the onset and gestation of pregnancy. Childbirth can proceed without complications and result in the birth of a healthy baby when amyloid protein accumulates in only one specific organ or tissue (intestinal wall, muscle tissue). In the generalized form, the prognosis for the mother and fetus is determined by the reserves of the affected organ and the duration of the disease.