- Advantages
- Doctors
- Contacts
- Licenses
Advantages
- The latest, constantly updated equipment
- Interest-free installments for all services
- Online consultations with an ENT doctor
- Visit of an ENT doctor to your home
- Friendly and qualified staff
- 24/7 ENT assistance
Pulmonary hypertension is a cardiovascular disease characterized by high blood pressure in the pulmonary artery. Pressure increases due to narrowing of the walls of the arteries leading from the heart to the lungs. This causes the heart to work harder to pump blood through the lungs. The extra effort eventually leads to weakening of the heart muscle and the development of serious heart failure.
Pulmonary hypertension can be life-threatening. Although some types have no cure, when diagnosed early, treatment for pulmonary hypertension reduces symptoms and improves quality of life.
The code for pulmonary hypertension according to ICD 10 is I27.0.
Symptoms of pulmonary hypertension
Signs of pulmonary hypertension appear slowly. They may go unnoticed for months or even years. Symptoms get worse (or more pronounced) as the disease progresses. These include:
- shortness of breath, first during physical activity, and then at rest;
- fatigue;
- dizziness;
- fainting;
- chest pressure or pain;
- swelling of the ankles, legs and eventually the abdomen (ascites);
- bluish color of lips and skin (cyanosis);
- increased pulse and heart rate.
Diagnostics
In addition to primary pulmonary hypertension, this syndrome is always a complication of a certain disease, which must be identified during diagnosis:
- analysis of medical history;
- analysis of the patient’s life history;
- general examination of the patient;
- electrocardiography;
- chest x-ray;
- Ultrasound of the heart;
- pulmonary artery catheterization;
- consultation with a cardiologist or pulmonologist.
Additional methods can be used that make it possible to establish the type and class of hypertension, and the patient’s tolerance to stress:
- spirometry and body plethysmography;
- blood gas analysis;
- assessment of the diffusion capacity of the lungs;
- chest tomography;
- angiopulmonography;
- general and biochemical blood test;
- detailed coalogram;
- Ultrasound of the abdominal organs;
- a 6-minute walking test, which allows you to understand the class of the disease and the patient’s tolerance to stress.
Degrees of pulmonary hypertension
Depending on the stage of development of the disease, its degrees are distinguished.
- Pulmonary hypertension stage 1. There are no clinical manifestations, physical activity is well tolerated, but in some areas of the pulmonary artery there are signs of circulatory disorders that can only be detected with a special examination.
- Pulmonary hypertension 2 degrees. Physical activity begins to bring discomfort. Worried about shortness of breath, weakness, dizziness. At rest I feel normal.
- Pulmonary hypertension grade 3. Even light physical activity causes severe discomfort.
- Pulmonary hypertension grade 4. All symptoms of pulmonary hypertension are felt even at rest; a person cannot lead a normal lifestyle.
Bronchopulmonary dysplasia
Bronchopulmonary dysplasia (BPD) is a polyetiological chronic disease of morphologically immature lungs, developing in newborns, mainly very premature infants, as a result of intensive therapy for respiratory distress syndrome and/or pneumonia. It occurs with primary damage to bronchioles and lung parenchyma, the development of emphysema, fibrosis and/or impaired alveolar replication; manifests itself as oxygen dependence at the age of 28 days of life and older, broncho-obstructive syndrome and symptoms of respiratory failure. It is characterized by specific radiographic changes in the first months of life and regression of clinical manifestations as the child grows [9].
Diagnostics
Diagnostic criteria: • artificial ventilation (ALV) in the first week of life and/or respiratory therapy with continuous positive airway pressure through nasal catheters (NCPAP - nose continuous positive airway pressure); • oxygen therapy more than 21% at the age of 28 days and older (oxygen dependence); • respiratory failure, broncho-obstructive syndrome at the age of 28 days and older, oxygen dependence that develops during oxygen therapy (ventilator, NCPAP); • interstitial edema alternating with areas of increased transparency of the lung tissue, fibrosis, band-like compactions on the chest x-ray. The diagnosis of “bronchopulmonary dysplasia” is valid as an independent diagnosis only for children under 3 years of age. At older ages, BPD is indicated only as a disease that has occurred in the anamnesis.
The main method of diagnosing the disease is considered to be radiological (x-ray and computed tomography of the chest). For an objective assessment of the severity of radiographic changes, G.V. Yatsyk, I.V. Davydova, O.V. Kustova in 2010 proposed and patented a scale on which each diagnostic sign was scored from 0 to 3 points, with a score of 0 points indicating the absence of the sign [10, 11]. The points were summed up: with a score from 1 to 5, a mild course was diagnosed, with a score from 6 to 10, a moderate course was diagnosed, and with a score from 11 to 15, a severe course of BPD was diagnosed (Table 2).
. Radiographic scale for assessing the severity of BPD, according to MSCT data.
Treatment
For children with established BPD, long-term (up to 6–12 months) basic therapy with corticosteroids is indicated; currently preference is given to inhaled glucocorticosteroids (budesonide) 500–100 mcg/day through a compression nebulizer. The criterion for discontinuation of budesonide is the absence of broncho-obstructive syndrome during the next acute respiratory viral infection (ARVI). In the acute period, the use of diuretics (furasemide 2 mg/kg), bronchodilators (Berodual 1 drop per 1 year of life through a nebulizer 3-4 times a day), cardiotrophics, and antibiotic therapy as indicated is indicated. If additional oxygenation is necessary, SaO2 saturation ≥ 92–95% is desirable in children who form cor pulmonale up to 96%. [11, 12].
Causes of pulmonary hypertension
Pulmonary hypertension is classified into five groups depending on the cause.
Group 1: pulmonary arterial hypertension.
Reasons include:
- Unknown cause (idiopathic pulmonary arterial hypertension).
- Genetic mutation (hereditary pulmonary arterial hypertension).
- Use of certain prescription diet drugs or illicit drugs.
- Heart problems from birth (congenital heart disease).
- Other conditions such as connective tissue diseases (scleroderma, lupus, etc.), HIV infection, or chronic liver disease (cirrhosis).
Group 2: pulmonary hypertension caused by left-sided heart disease.
Reasons include:
- Left-sided heart valve disease.
- Failure of the lower left chamber of the heart (left ventricle).
Group 3: pulmonary hypertension caused by lung disease.
Reasons include:
- Chronic obstructive pulmonary disease.
- Pulmonary fibrosis.
- Obstructive sleep apnea.
- Prolonged exposure to high altitude in people who are at higher risk of pulmonary hypertension.
Group 4: thromboembolic pulmonary hypertension caused by chronic blood clots.
Reasons include:
- Chronic blood clots in the lungs (pulmonary embolism).
- Other blood clotting disorders.
Group 5: secondary pulmonary hypertension caused by other diseases.
Reasons include:
- Blood diseases.
- Inflammatory diseases.
- Metabolic disorders, including glycogen storage disease.
- Kidney diseases.
- Tumors pressing on the pulmonary arteries.
- Eisenmenger syndrome.
Pulmonary hypertension in children can be of all the forms described above. However, the most common pulmonary arterial hypertension that develops as a result of heart defects and idiopathic pulmonary hypertension occurs in them.
Medical Internet conferences
Primary pulmonary hypertension (PPH) - Ayersa's disease - is a rare, severe, progressive disease of unknown etiology and is characterized by a poor prognosis [1, 2]. Pulmonary arterial hypertension (PAH) is diagnosed when the mean pressure in the pulmonary artery increases to or more than 25 mmHg. Art. at rest and 30 mm Hg. Art. under load [3].
According to the literature, the incidence of idiopathic pulmonary hypertension is 1–1.7 cases per million people [2], and the disease is almost 2 times more common in women than in men. The average age of patients with this diagnosis is 36.4 years [1]. Survival statistics for patients with PLH are not reassuring - in the absence of intensive treatment, the average life expectancy is about 3 years from the moment of diagnosis [2].
Despite the active study of the problem, the etiology of PLH remains unclear, but researchers suggest an important role of genetic disorders in the development of the disease [4]. In particular, the development of PLH is associated with a mutation of the BMPR2 gene, localized on the second chromosome, but the pathogenetic relationship between this disorder and PLH has not been discovered [11].
Despite the fact that the factors leading to the triggering of pathological changes in the pulmonary vessels have not been precisely established, today researchers consider the theory of endothelial dysfunction (ED) to be the main one in the etiology and pathogenesis of this disease, which indicates that damage and dysfunction of the endothelium of the pulmonary vessels leads to the development of vasoconstriction and remodeling of the vascular bed. In pathogenesis, four main pathophysiological phenomena are distinguished: vasoconstriction, reduction of the pulmonary vascular bed, decreased elasticity of the pulmonary vessels, their obliteration due to thrombosis and proliferation of smooth muscle cells (SMCs). These processes are induced by an imbalance in the production of vasoactive mediators that occurs during ED, which consists of increased synthesis of vasoconstrictor substances (thromboxane A2, endothelin-1) by endothelial cells and a deficiency in the production of vasodilating substances (NO, prostacyclin). Disruption of the voltage-gated potassium channels of SMCs also plays a role, which causes vasoconstriction and proliferation of SMCs due to increased intracellular calcium ion content. A key role in the development and progression of PAH is played by NO deficiency, which promotes chronic vasospasm, cell proliferation, and intravascular thrombus formation [5].
Thus, a vicious circle is closed - progressive damage to the endothelium leads to vascular remodeling [11].
Pathological processes in PAH affect all layers of the vascular wall, various types of cells (endothelial, SMC, fibroblasts). Structural disorders begin with muscular arteries and pulmonary arterioles. The process begins with hypertrophy and muscularization of the medial tunic of arterioles. In addition, migration of SMCs into the intima of the vessel is observed, in which active proliferation of cellular elements occurs. Initially, intimal thickening is reversible, but soon irreversible changes develop - fibrosis and fibroelastosis. In some cases, concentric intimal fibrosis completely closes the lumen of the vessel. In this case, plexogenic and pleximorphic structures can be determined, which, as a rule, arise proximal to the site of obstruction. The adventitia exhibits increased production of extracellular matrix, including collagen, elastin, fibronectin, and tenascin. The secretion of mediators with a pronounced vasoconstrictor effect transforms the state of the vascular bed from the usual anticoagulant to procoagulant and contributes to the development of thrombosis [10, 11].
Modern functional studies using various non-invasive methods for assessing the state of peripheral blood flow do not confirm the generalization of endothelial dysfunction in patients with PLH, however, an increased constrictor ability of the SMC is noted [6].
An increase in vascular resistance in the pulmonary circulation (PCC) initiates the occurrence of myocardial hyperdynamia syndrome with the development of right ventricular (RV) hypertrophy. This phenomenon is due to the physiological response described by Starling, which consists of an increase in the force of cardiac contraction in response to load by volume or vascular output resistance [7]. Cor pulmonale (CH) develops with primary or secondary PAH resulting from dysfunction of pulmonary vascular endothelial cells and structural and functional changes in the pulmonary vessels [8]. There are several stages in the development of the pulmonary heart. In the initial stage, there is an increase in pulmonary vascular resistance, which is a consequence of PAH. In the second stage, changes in the pancreas are detected in response to an increase in pulmonary arterial pressure. The third stage consists of pronounced hypertrophy and dilatation of the RV, changes occur in the vascularization of the latter, and right ventricular heart failure (RVHF) may be observed [9].
During the formation of LS, changes in the myocardium are observed in the form of hypertrophy, dystrophy, atrophy and necrosis of pancreatic cardiomyocytes. Atrophic-sclerotic processes can be observed in the muscle fibers of the pancreas, and pronounced endocardial fibroelastosis can be observed in the right atrium and pancreas. Less common is dilatation of the left ventricular cavity [12].
Diagnosis of PLH is difficult because patient complaints are nonspecific and may resemble congenital heart disease. In most cases, clinical manifestations develop after the development of irreversible changes in the lungs. Weakness, fatigue, shortness of breath, hemoptysis, anginal pain in the heart area, dizziness, and abdominal discomfort are noted. Upon examination, pulsation of the neck veins, hepatomegaly, peripheral edema, and ascites are detected. Palpation is determined by pulsation in the fourth intercostal space to the left of the sternum, enlargement of the right ventricle. An increase in the second tone in the second intercostal space on the right is auscultated [13].
Conservative treatment of PLH involves, first of all, the prescription of calcium antagonists, anticoagulant therapy, and the use of diuretics for PHF. Sildenafil citrate (the active ingredient of Viagra) has the same vasodilating abilities in both the vessels of the genitals and the lungs, and therefore was approved in the USA as a drug for the treatment of PLH [4]. Surgical treatment is also possible - transplantation of the heart-lung organ complex [11]. However, complete elimination of PLH is currently not possible; treatment is supportive.
Persistent pulmonary hypertension of newborns (PPHN) is included in a separate category due to the presence of characteristic pathogenetic and clinical and morphological aspects associated with the characteristics of the fetal blood circulation and distinguishing it from PHH. PPHN is a disease characterized by abnormally increased pressure in the pulmonary vessels and, as a consequence, right-to-left shunting of blood through the foramen ovale and (or) patent ductus arteriosus in the absence of other heart defects. Among the variants of PPHN, in turn, there are idiopathic (primary) and complicating various neonatal cardiopulmonary complications (secondary) [14]. PPHN occurs in 0.2-0.3 cases per 1000 newborns, but the idiopathic variant accounts for up to 20% of all cases of PPHN [15].
The leading etiology of idiopathic PPHN is prolonged intrauterine hypoxia, which leads to vascular remodeling, which is expressed in vascular smooth muscle hyperplasia, spreading to intralobular arteries, which leads to increased vascular resistance [15].
In healthy newborns, the activity of vasoactive substances changes in response to numerous stimulations that occur during birth, including increases in partial pressure of oxygen and changes in blood pH [14]. The main pathogenetic link of idiopathic PPHN is also considered to be an imbalance in the synthesis of vasoconstrictor and vasodilating substances [15].
Thus, primary pulmonary hypertension is a rare and severe disease with an extremely poor prognosis. However, the accumulated clinical experience provides opportunities to discover the mechanisms of its pathogenesis and develop new diagnostic and treatment methods that can not only prolong the life of patients, but also improve its quality. In this regard, relevant .
Clinical case
From the anamnesis it is known that the boy E.D. was born from the fifth pregnancy (second birth), occurring against the background of the threat of miscarriage, edema of pregnant women, low placentation, partial progressive abruption of the low-lying placenta, a burdened obstetric and gynecological history, chronic intrauterine fetal hypoxia. Preterm birth at 32–33 weeks gestation was resolved by emergency cesarean section.
The newborn was in serious condition with an Apgar score of 4-5-6-6 points. The severity of the condition was due to respiratory disorders, neurological symptoms due to prematurity and morphofunctional immaturity. Anthropometric data of the child at birth: body weight 2530 g, height 47 cm, chest circumference 31 cm, head circumference 33 cm.
From birth he was on a ventilator. At the maternity hospital stage, respiratory, infusion, antibacterial and hemostatic therapy was carried out. For the purpose of further examination, treatment and nursing, the boy was transferred by an air ambulance team from the maternity hospital to the neonatal intensive care unit of the Saratov Regional Children's Clinical Hospital (SODKB).
Based on the examinations performed, the main competing clinical diagnosis was made: 1. Congenital pneumonia. 2. Cerebral ischemia grade 3, acute period. Oppression syndrome. Complications: multiple organ failure syndrome (cerebral edema, stage 3 coma; stage 3 respiratory failure; stage 3 acute cardiovascular failure; acute renal failure in the stage of oligoanuria; edema syndrome; hemorrhagic syndrome; pulmonary hypertension). Concomitant diseases: perimembranous subtricuspid interventricular septal defect 0.24 cm; patent ductus arteriosus: 0.30cm; open functioning foramen ovale: 0.42 cm, left-right reset. Background diagnosis: neonatal respiratory distress syndrome; prematurity 32-33 weeks.
Under the conditions of the therapy (inotropic support, surfactants, increasing ventilation parameters to the maximum possible), the child’s condition remained severe with deterioration dynamics. Against the background of progressive multiple organ failure, asystole was noted, and the biological death of the child was stated.
When opening the chest cavity, the usual arrangement of organs was noted, but the lungs completely filled the pleural cavity. The shape of the heart is cone-shaped, the location is normal. The size and location of the thymus gland are unremarkable. In the soft tissues there are bright red hemorrhages measuring up to 1 cm.
When examining the respiratory tract, swollen pink palatine tonsils of normal size were noted. A small amount of mucous content is detected in the lumen of the larynx, trachea and bronchi. The mucous membrane of these organs is edematous, smooth along its entire length. The leaves of the parietal and visceral pleura are smooth, pale pink. The lungs are dark red in color, dense in consistency, the lung tissue is homogeneous when cut, abundantly saturated with blood; when pressed, liquid blood and edematous fluid are released. The weight of the left lung is 40 grams, the right lung is 37 grams. Pieces cut from the lungs sink when immersed in water. Histological examination of the lungs revealed focal accumulations of erythrocytes and forming hyaline membranes in the lumens of the alveoli; in the vessels, hypertrophy of the muscular layer of arteries and veins was noted by more than 25%, intimal proliferation with fibrosis, as well as thrombotic masses were detected.
An examination of the circulatory system revealed a small amount of yellowish transparent fluid in the cardiac membrane. The pericardial leaves are smooth and shiny. Heart weighing 20 g. Myocardium pink-red, elastic consistency. The valvular and parietal endocardium throughout is thin, translucent, shiny, with a grayish tint. The thickness of the myocardium of the left ventricle is 0.7 cm, the right one is 0.3 cm (normal is 0.2 cm). The perimeter of the pulmonary trunk above the valves is 2.2 cm, of the aorta - 1.9 cm. The intima of the aorta is smooth, shiny, reddish-yellow. There is dark liquid blood in the cavities of the heart and in large vessels. The heart valves are developed correctly and not altered. Interventricular communication measuring 0.2 cm; the oval window measuring about 0.3 cm is open; open arterial duct measuring 0.3 cm. Histological examination revealed dystrophy and hypertrophy of cardiomyocytes.
When opening the cranial cavity, it was discovered that the dura mater was thin, with a pearlescent sheen. The soft meninges are swollen and full of blood. The convolutions of the brain are flattened, the furrows are smoothed. The lateral ventricles are slightly increased in size and contain clear, bright yellow cerebrospinal fluid. In the cavity of the right ventricle there is a small blood clot of bright red color with a volume of about 5 ml. The choroid plexuses are full-blooded, with transparent contents. The white and gray matter of the cerebral hemispheres are distinguishable; dark red dots are identified on the surface of the sections, easily washed off with water. The rest of the brain structures are without visible changes. Brain weight 265 grams. Histological examination revealed uneven blood supply to the cerebral vessels.
When the abdominal cavity was opened, about 5 ml of yellow liquid was released. The loops of the small intestine in the lower parts are dark red in color and slightly increased in size; the intestinal vessels are full of blood. The mucous membrane of the small intestine has a smoothed pattern, dark red in color. There is hemorrhagic content in the lumen of the lower parts of the small intestine. The liver, weighing 105 grams, is red in color and has a normal structure on the surface and in the cut. Histological examination of the liver revealed degeneration of hepatocytes and congestion of the hepatic sinuses.
When opening the kidneys, it was noted that on the section the cortical substance was bluish-red in color, and the medulla was dark red; parenchyma of dense consistency; The mucous membrane of the pelvis is smooth, clean with isolated small hemorrhages. The mucous membrane of the ureters and bladder is without features. Histological examination of the kidneys revealed dystrophy of the epithelium of the convoluted renal tubules, as well as uneven blood supply to the vessels.
During the autopsy of the remaining organs and systems, no significant features were identified.
Based on the autopsy and histological examination, a pathological diagnosis was made - primary pulmonary hypertension; complications of the underlying disease – myocardial hypertrophy of the right ventricle of the heart; secondary hyaline membranes; hemorrhages in the lungs; parenchymal dystrophy of internal organs.
Discussion
In this case, PPH in a newborn was combined with a patent foramen ovale and patent ductus arteriosus, as well as a perimembranous ventricular septal defect, which led to significant changes in hemodynamics in the neonatal period. The consequence of prolonged hypoxia was ischemic damage to various organs, the development of parenchymal degeneration and multiple organ failure, which determined the severity of the patient’s condition. The immediate cause of death of the patient was respiratory failure.
Conclusion
Thus, PPH, a severe progressive disease that is difficult to treat, is complicated in newborns by hemodynamic features and inevitably leads to death. Studying the etiology and pathogenetic mechanisms of idiopathic pulmonary hypertension will allow us to find new ways to help patients suffering from this disease.
Risk factors
As you get older, your risk of developing chronic pulmonary hypertension increases. The disease is more often diagnosed in people aged 30 to 60 years. However, the idiopathic form is more common in young people.
Other factors that may increase the risk of pulmonary hypertension include:
- family medical history;
- excess weight;
- bleeding disorders or a family history of blood clots in the lungs;
- exposure to asbestos;
- genetic disorders, including congenital heart defects;
- life at high altitude;
- using certain drugs for weight loss;
- use of illegal and narcotic drugs;
- use of selective serotonin reuptake inhibitors, used to treat depression and anxiety.
Treatment
If the disease is pulmonary hypertension, treatment includes non-drug and medicinal methods. Non-drug methods:
- limiting fluid intake to 1.5 liters per day, reducing the amount of table salt in the diet;
- oxygen therapy, which has a positive effect on the restoration of central nervous system functions and helps eliminate acidosis, while breaking the chain of action of the syndrome;
- The patient should avoid symptoms of shortness of breath, pain, and fainting. To do this, you need to control physical activity;
- Climbing to heights must be avoided.
Medication methods:
- calcium antagonists, which can not only change the frequency of heart contractions, but also reduce spasms of blood vessels in the pulmonary circulation, relax muscles, and increase the stability of the heart muscle relative to hypoxia;
- Diuretics are drugs that remove excess fluid from the body and reduce blood pressure. When treated with diuretics, electrolyte composition and blood viscosity should be monitored;
- ACE inhibitors. Reduce blood pressure, reduce stress, dilate blood vessels;
- nitrates - reduce stress by dilating the veins of the extremities;
- antiplatelet agents - help reduce the level of adhesion of red blood cells and platelets and their adhesion to the lining of internal vessels;
- direct-acting anticoagulants - drugs that prevent the formation of fibrin and prevent the development of blood clots;
- indirect anticoagulants. Drugs in this group act on the formation of factors that provoke blood clotting, thereby reducing it;
- antagonists for endothelin receptors. These are powerful agents that dilate blood vessels;
- drugs that improve bronchial patency - bronchodilators. They improve ventilation in the lungs, especially in those conditions that are accompanied by reversible bronchospasms;
- antibiotics (in the presence of bronchopulmonary infection);
- nitric oxide - used for inhalation to dilate blood vessels;
- phosphodiesterase type 5 inhibitors, which help improve blood flow through the vessels and dilate them;
- prostaglandins are powerful vasodilators, which also have many additional effects - antiaggregation, antiproliferative, cytoprotective.
Surgery:
- atrial septostomy - an artificial opening is created between the left and right atria, which helps reduce the level of pressure in the pulmonary artery;
- thromboendatherectomy - removal of blood clots from blood vessels through surgery;
- transplantation of lungs or lungs at the same time as a heart. Used for severe pulmonary hypertension and concomitant complex cardiac diseases.
Complications
The following complications may accompany the disease:
- the appearance of right ventricular failure of the heart, in which the right parts of the heart cannot cope with the load. As a result, the patient's condition becomes even worse;
- thrombosis of the pulmonary arteries. This condition seriously threatens the patient’s life and is caused by mechanical blockages of the pulmonary artery trunk;
- heart rhythm disturbances - atrial fibrillation and tremors;
- hypertensive crises in the arterial system of the lungs, which manifest themselves as attacks of pulmonary edema: a sharp increase in suffocation, severe cough, sputum, blue discoloration of the skin.
- in general, with a syndrome such as pulmonary artery hypertension, the patient’s quality of life greatly deteriorates and the disease can cause a shortened life span.
With a disease such as pulmonary hypertension, the prognosis may not always be optimistic. A fatal outcome is likely as a result of acute or chronic failure of the heart and lungs, as well as thromboemblyolia of the artery in the lungs.
Prevention
In order to prevent problems such as pulmonary hypertension, recommendations are as follows:
- healthy lifestyle, giving up bad habits, especially smoking. Curing the underlying disease that caused pulmonary hypertension. (If we are not talking about primary hypertension);
- dispensary observation of people who have been diagnosed with bronchopulmonary diseases and therapeutic measures have been taken in a timely and competent manner;
- People with pulmonary hypertension need regular physical activity. These are low-intensity exercises, such as moderately brisk walks in the air if tolerated. Strong activity is dangerous and therefore contraindicated;
- elimination of intense psycho-emotional stress - stress, conflict situations, since they also have a direct impact.
Diagnosis of pulmonary hypertension
Pulmonary hypertension is difficult to diagnose at an early stage, so it is rarely detected during a routine physical examination. Even the symptoms of more advanced pulmonary hypertension are often similar to other heart and lung diseases.
The doctor performs a physical examination and asks questions about your medical and family history. After that, medical tests are prescribed that will help make a diagnosis of pulmonary hypertension and determine its cause. Tests for pulmonary hypertension may include:
- blood tests;
- chest x-ray;
- electrocardiogram (ECG);
- an echocardiogram (sometimes while exercising on a stationary bike or treadmill);
- right heart catheterization;
- computed tomography (CT);
- magnetic resonance imaging (MRI);
- pulmonary function test (measures how much air the lungs can hold and the flow of air into and out of the lungs);
- polysomnogram (during sleep);
- histological examination of biopsy material from an open lung;
- genetic tests (if the test result is positive, the doctor may recommend that other family members be tested as well).
Clinical observation
In order to focus the attention of pediatricians, pediatric cardiologists and pulmonologists on this problem, we present an interesting clinical case of PAH against the background of a combination of BPD and congenital heart disease in a child with ELBW at birth.
Child A., female, from the 1st pregnancy, which proceeded with the threat of termination in the first and second half, was born from 1 premature birth at the 26th week. gestation, by Caesarean section, with a weight of 870 g and a length of 34 cm, with an Apgar score of 4/6 points.
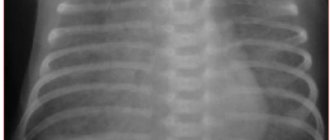
From the first hours of her life, the girl had a serious condition with the manifestation of respiratory distress syndrome. Resuscitation measures were carried out using mechanical ventilation, oxygen therapy, surfactant replacement therapy, and incubation regimen. On the 2nd day of life, a hemodynamically significant patent ductus arteriosus (PDA) was detected, 3 doses of ibuprofen were administered intravenously according to the usual regimen, without a positive effect. Over time, the manifestations of heart failure increased. In this regard, the child received cardiac glycosides, dopaminomimetics, β-adrenergic agonists (dopamine, digoxin, dobutamine). With this therapy, the condition stabilized somewhat, although it remained serious. Persistent respiratory failure of the 3rd degree was noted, from the 3rd day anemia (Hb 102 g/l), hematuria were noted, fine rales and crepitus were heard on auscultation in the lungs on both sides. On the 8th day of life, radiological changes were revealed in the form of a bilateral decrease in pneumatization due to gentle infiltrative shadowing in both lungs with an increase in the bronchovascular pattern due to the interstitial component (Fig. 1), leukocytosis (21.0 × 109/l), there was a negative dynamics in blood tests on the 1st week. life (increase in leukocytosis, ESR, decrease in hemoglobin).
A diagnosis was made: Neonatal pneumonia, bilateral, focal, severe, infiltration stage. DN III degree. Microbiological culture of throat and endotracheal aspirate revealed Klebsiella pneumoniae
. In addition, in a child with a burdened obstetric history, arteritis and phlebitis of the umbilical vessels, anemia, and pneumonia, signs of SIRS (thrombocytopenia, impaired thermoregulation, inflammatory changes in the blood) were recorded, which suggested management as being at risk of neonatal sepsis. Due to many diseases, the girl received complex therapy (including antibacterial) and continued to be on mechanical ventilation for a long time (FiO2 0.45; f 40 in 1 min; pi. 19 mm Hg; SaO2 90–92%) . Periodically, attempts were made to reduce the parameters of mechanical ventilation, however, due to persistent hypercapnia (pCO2 55 mm Hg) and hypoxia (pO2 24 mm Hg), acidosis, according to the acid-base composition of the blood (lactate 1.1 mmol/ l; cHCO3 38.2 mmol/l; BE 12.3 mmol/l), low levels of oxygen saturation, it was not possible to extubate the child.
From the 28th day of life, in accordance with a set of criteria [14], a diagnosis was made: Bronchopulmonary dysplasia of prematurity, a new form, severe. The main point for making the diagnosis was the need for oxygen concentration of more than 21% on the 28th day of life, oxygen dependence at 36 weeks. postconceptional age indicated severe BPD. A computed tomogram revealed the main signs of BPD—fibrous cords, ground glass, and bullous swellings (Fig. 2). For the disease, the child received therapy with inhaled glucocorticosteroid (ICS) budesonide at a dose of 1.0 mg/day, muco- and bronchodilator therapy through a nebulizer (ambroxol, fenoterol, ipratropium bromide).
At the age of 2.5 months. Life diagnosis confirmed: Congenital heart defect (CHD): PDA. On ECHO-CG: PDA 3.5 mm with blood discharge into the pulmonary artery with a gradient of 4 mm Hg. Art., aneurysm of the interatrial septum (ASA) and a patent foramen ovale 3.5 mm with right-to-left shunting, high PAH 89 mm Hg. Art. with expansion of the right atrium (RA) (Fig. 3).
The ECG showed signs of RA overload. X-ray: signs of enlargement of the right chambers of the heart. In the hemogram: anemia Hb 90 g/l, Er 3.88 × 1012/l, CP 0.9, leukocyte count without signs of inflammation. Blood glucose was 5.0 mmol/L, creatinine 51.5 mmol/L, ALT 0.5 µmol/L, AST 0.94 µmol/L. At the age of 3.5 months. An operation for PDA (ligation) was performed at the Federal Center for Cardiovascular Surgery (Astrakhan). The early postoperative period passed without complications.
For the identified pathology of congenital heart disease + PAH, the child received complex therapy, including oxygen therapy, diuretics (spironolactone under the control of creatinine and potassium), sildenafil (at a dose of 3.5 mg × 4 times / day), digoxin at a maintenance dose (10 mg/kg /day), dobutamine (1.5 mcg/kg/min). The child's condition stabilized, shortness of breath decreased, appetite improved, and the girl began to gain body weight of 350 g or more per week.
The diagnosis was made: Deep prematurity (26 weeks of gestation). BPD of prematurity, a new form, severe severity, respiratory failure of II degree. Congenital heart disease: PDA, stage IIA heart failure, FC II, minor cardiac anomaly: patent foramen ovale. Consequences of hypoxic damage to the central nervous system, depression syndrome. Early anemia of prematurity of moderate severity. Retinopathy of the retina of both eyes, stage III, active phase.
Further, against the background of treatment and positive clinical and laboratory dynamics (increase in weight and height indicators, absence of inflammatory changes in the hemogram, negative CRP and PCT indicators, absence of pathological deviations of other biochemical indicators, improvement in SaO2 indicator: 94–95%) on the 65th day of life, the child was extubated and transferred to spontaneous breathing. Inhaled therapy for severe BPD with ICS (budesonide) was continued at the same volume.
Against the background of stabilization of the condition at the age of 5 months. Surgical treatment of retinopathy of prematurity was carried out (at the St. Petersburg State Pediatric Medical University of the Ministry of Health of Russia, Department of Pathology of Newborns and Infants). After the operation, the dynamics of the child remained persistent respiratory failure (RR 42–44, with anxiety up to 85 per minute; SaO2 90–94%), which was associated with prolonged oxygen support, the total duration of receiving the air-oxygen mixture was about 6 months.
On an outpatient basis, the child was observed by a pediatrician, pulmonologist, and cardiologist with a diagnosis of Bronchopulmonary dysplasia of prematurity, a new form, severe, complicated by secondary high pulmonary hypertension (89 mm Hg). Condition after correction of congenital heart disease (ligation of the PDA, AMP, patent foramen ovale, ectopic chord in the cavity of the left ventricle). Paresis of the left vocal cord. Consequences of hypoxic damage to the central nervous system. Delayed physical and motor development. Stage III retinopathy of the retinas of both eyes, active phase (surgical treatment: transpupillary laser coagulation of the retina). Moderate anemia of prematurity. Protein-energy deficiency of the third degree.
During the first year of life, the child had extremely low physical development, disharmonious due to weight deficiency (at 1 year, weight 6.0 kg and body length 58 cm) -6.2SD and -3.2SD in height and weight, respectively [17]. Noteworthy were increased weakness, fatigue, dry cough during the day, periodic stridor breathing, shortness of breath at rest of a mixed nature from 65 respiratory movements per minute at rest, remission and up to 82 per minute during periods of exacerbation of BPD. Oxygen saturation in peripheral blood by pulse oximetry was 90–95%. During the observation period, the most severe deterioration of the condition was observed twice at the age of 6 months. and 8 months, which was associated with respiratory infections.
The girl received basic therapy for BPD with the drug budesonide at a dose of 1.0 mg/day from the moment of diagnosis continuously for 6 months, then 0.5 mg 1 time/day for 6 months. During the period of exacerbation of the disease, the dose of ICS was increased to 2.0 mg/day, and ipratropium bromide was used at a dose of 0.075 mg (up to 6 drops) to relieve shortness of breath.
During outpatient treatment for pulmonary hypertension, a course of sildenafil was continued at a dose of 4 mg/kg/day × 4 times/day for 2 weeks, then 1 mg/kg/day for 6 months, spironolactone 2 mg/kg × 2 rubles/day
During observation for 1 year, systolic pressure in the pulmonary artery (SPAP) had a positive trend. The ECG recorded sinus rhythm, pacemaker migration, the electrical axis of the heart was sharply deviated to the right, â=+150°, heart rate 120–150 per minute, PQ 0.1. ECHO-CG shows: the condition after ligation of the PDA, the duct is hermetically closed, the borderline dimensions of the right chambers of the heart (right ventricle 1.77 cm; RA 1.76 cm). Global contractility of the left ventricle is not impaired. Emission fraction SF 60%. Ectopic chord in the cavity of the left ventricle. Mitral regurgitation grade 0–1. Tricuspid regurgitation of the first degree. Pulmonary artery 1.46 cm, MPAP 46 mm Hg. Art. The pericardium is without features. The maximum pressure gradient at Ao is 8.1 mm Hg. Art. O2 saturation 94%.
During treatment, a positive dynamics of MPAP was noted from 89 to 46 mm Hg. Art., clinical signs of PAH have decreased significantly, signs of BPD and the consequences of hypoxic damage to the central nervous system, delayed physical and psychomotor development, and correction of congenital heart disease remain.
Forecast
The prognosis for life and recovery with pulmonary hypertension depends on the form and stage of the disease, the timeliness and adequacy of the therapy. When using modern treatment methods, the mortality rate of patients with chronic forms of the disease is 10%. The five-year survival rate of patients with primary pulmonary hypertension varies from 20 to 35%.
The overall prognosis is influenced by the following factors:
- The degree of pulmonary hypertension by pressure - with a decrease in pressure in the pulmonary artery, the prognosis will be favorable, with an increase in pressure by more than 50 mm Hg. Art. – unfavorable. A patient diagnosed with secondary pulmonary hypertension may be more likely to have a favorable prognosis;
- An increase in symptoms of the disease or a decrease in their severity;
- Improvement or deterioration of the patient's condition during therapeutic treatment.
When pulmonary hypertension develops in newborns, the prognosis depends on how long the doctor identifies the problem. In most cases, it takes up to three days to make a diagnosis, after which doctors begin to implement a set of therapeutic measures.