General information
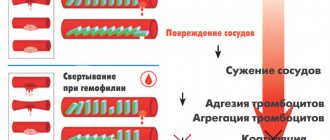
The blood coagulation system is a complex sequential cascade of reactions occurring in the body that is aimed at stopping bleeding. The coagulation process is an important protective reaction of the body, thanks to which a constant volume of circulating blood is maintained. homeostasis system involves many components, the main ones of which are shown in the figure below.
Clotting factors are in an inactive state in the blood. If a blood vessel is injured, the coagulation process begins and all factors are sequentially activated and ensure the formation of a clot. The direct coagulation process itself is associated with the conversion of the fibrinogen protein (factor I) into insoluble fibrin.
Coagulopathy is a disease, or rather a group of diseases or conditions, which are based on a blood clotting disorder. The clinical sign of coagulopathy is bleeding. Acquired coagulopathies are the most common syndromes. In this case, poor blood clotting can be caused by pathology of different parts of the coagulation system: fibrin , platelets or blood clotting factors . If any part of this system does not function or is missing, the person will have prolonged bleeding and develop a critical condition. Among coagulopathies there are congenital conditions and diseases ( hypofibrinogenemia (factor I deficiency), afibrinogenemia , hemophilia A , von Willebrand disease , hemophilia B ) and acquired syndromes that occur in various septic conditions and diseases of the kidneys and liver.
Hypofibrinogenemia
In late pregnancy, fibrinogen levels are 3-6 g/l. When coagulation is activated, this high level of fibrinogen may sometimes play a protective role against clinically significant hypofibrinogenemia. To ensure clinical coagulation, the fibrinogen level should not be less than 1.5 g/l. In the presence of significant hypofibrinogenemia, a whole blood blood clot in a glass tube may initially be soft, although its volume does not decrease significantly. But after a while (about 30 minutes) it becomes small as red blood cells and fluid are released from it.
Fibrin and fibrinogen derivatives . Fibrinogen degradation products in blood serum are a sensitive marker of coagulopathy, and their determination is the basis of many test systems (monoclonal antibodies). An increase in the content of fibrinogen degradation products indicates severe consumption coagulopathy.
Pathogenesis
The pathogenesis of hemorrhagic syndrome includes:
- damage to vascular endothelial cells by inflammatory mediators or endotoxin ;
- activation of protein C , which inhibits FV and FVIII and suppresses the synthesis of plasminogen activator inhibitor (the latter promotes the transition of plasminogen to plasmin, which breaks down the fibrin of the blood clot);
- activation of fibrinolysis, which plays a role in the development of non-stop bleeding and depletion of f I, II, V, VIII, XIII;
- accumulation in the blood of metabolites that have an anticoagulant effect.
In the pathogenesis of uremic thrombocytopathy, platelet deficiency is important, which is associated with the action of toxic plasma metabolites. In addition, patients with uremia undergo an extracorporeal circulation procedure, in which platelet dysfunction occurs due to their interaction with the tubes and membranes of the device. In this case, platelets are activated and release granules (platelet degranulation). Platelet dysfunction causes bleeding so severe that platelet transfusion is required.
Drug-induced thrombocytopenia is associated with the interaction of the drug (or its metabolite) and a platelet membrane glycoprotein. As a result of this interaction, an immunogenic complex - a glycoprotein-drug . Altered platelets are removed from the bloodstream by RPE cells. With drug-induced thrombocytopenia, the level of IgG and platelet-bound antibodies to the drug increase. Idiopathic purpura based on the production of antibodies against viral antigens. Platelets are damaged by adsorbing a viral antigen or a virus-antibody immune complex on their membrane.
Classification
Coagulopathies are divided into hereditary and acquired.
- Hereditary forms are associated with genetically determined changes in the walls of blood vessels, abnormalities of platelets and plasma blood factors. Hereditary coagulopathies include hemophilia A , von Willebrand disease , hemophilia B , and deficiency of various clotting factors.
- Acquired forms are most often associated with vascular damage of various etiologies (immune, toxicoinfectious and dysmetabolic), platelet damage, pathology of coagulation factors and a combination of all these factors.
The following types of acquired coagulopathy are distinguished:
- Disorders of platelet hemostasis . These include thrombocytopenia of various origins (associated with decreased platelet production, associated with increased platelet destruction, caused by non-immune causes and immune ones - idiopathic thrombocytopenic purpura), HELLP and hemolytic-uremic syndrome, thrombotic thrombocytopenic purpura, thrombocytopathies. With thrombocytopenia, the platelet germ may be primarily affected, platelets can be redistributed and accumulate in the spleen, and there will be an insufficient number of them in the blood. Also, platelets can be destroyed in large quantities (with lupus erythematosus and thrombocytopenic purpura). In addition, platelets can be consumed in large quantities during the formation of blood clots (for example, in DIC syndrome). Thrombocytopathies are characterized by the production of abnormal platelets whose function is impaired. An example of thrombocytopathy is von Willebrand disease and Glanzmann thrombasthenia .
- Types of coagulation disorders. This group includes hemostasis disorders associated with an overdose of anticoagulants ( heparin , warfarin ), hemodilution coagulopathy , vitamin K-dependent (with impaired liver function, poor absorption of vitamin K, taking certain medications).
- Coagulation-platelet (mixed) disorders that developed against the background of liver and kidney failure (hepatic and uremic coagulopathy).
- DIC syndrome is distinguished separately.
Fibrinogen plays an important role in the blood coagulation system . Normally its content is 2-4 g/l. Patients often have hypofibrinogenemia —a decrease in fibrinogen levels. This condition can be hereditary, but more often acquired, due to insufficient formation of this liver protein when it is damaged or its increased dissolution (fibrinolysis). In this condition, blood clotting slows down and as a result, a loose, disintegrating clot is formed. Hypofibrinogenemia manifests itself through the formation of bruises with minor trauma and various bleeding.
However, in most patients, decreased fibrinogen levels do not manifest themselves in any way. A decrease in the level of this protein is observed in cirrhosis , liver necrosis , bone marrow metastases leukemia , shock , anemia , eclampsia , premature placental abruption, complicated childbirth, and sepsis. The cause of acute hypofibrinogenemia is intravascular coagulation when fibrinogen is intensively consumed. Many patients with low fibrinogen levels do not require treatment. Heavy menstruation in women is prevented by hormonal agents and antifibrinolytic drugs.
Afibrinogenemia is a complete absence of fibrinogen in the blood with a normal platelet level. This is a genetically determined disease, but is very rare. In this condition, any injury leads to bleeding, hematomas, and hemorrhages in the joints. Dental manipulations and operations are accompanied by significant blood loss. Children rarely survive to adulthood. In the treatment of this pathology, replacement therapy is used: administration of fibrinogen , cryoprecipitate and fresh frozen plasma .
Hereditary coagulopathies: hemophilia and von Willebrand disease
The human blood coagulation system is a multicomponent and extremely complex mechanism that plays a critical role in protecting the entire body.
This mechanism is represented by three links: vascular, platelet and plasma coagulation. Diseases associated with the inability of blood coagulation factors to ensure the normal process of clot formation constitute a large group of coagulopathies. This group of diseases is represented by hereditary coagulopathies and many acquired forms of coagulopathies, which are the result of other diseases: cirrhosis and liver cancer, autoimmune (systemic lupus erythematosus, immune thrombocytopenic purpura, hemorrhagic vasculitis, etc.), infectious diseases, toxic effects of drugs and poisons, hereditary metabolic diseases.
Hereditary diseases of hemostasis
Hemophilia and von Willebrand disease are the most common hereditary diseases of the plasma hemostasis system. Von Willebrand factor and factor VIII in the blood plasma are presented in the form of a molecular complex, while von Willebrand factor plays a protective role for factor VIII, protecting it from destruction by protein C.
This is why, in the absence of von Willebrand factor, factor VIII levels can be significantly reduced. Thus, despite the differences between these diseases, they exhibit a certain molecular biological synergy. In this regard, it is advisable for practicing physicians to consider two diseases that account for more than 90% of hereditary coagulopathies caused by a deficiency or functional failure of fiery blood coagulation factors - VIII, IX (hemophilia A or B, respectively), or von Willebrand factor (von Willebrand disease).
Factor VII deficiency is also extremely rare. Hypoproconvertinemia manifests itself as a hemorrhagic syndrome, depending on the severity of factor VII deficiency. In severe cases, hemarthrosis, hematomas are observed, and in women, menorrhagia.
Both occur and are diagnosed infrequently
In general, hereditary coagulopathies are uncommon and even less commonly diagnosed. There are two main reasons for this: low prevalence, often associated with a subclinical course of the disease (in von Willebrand disease) and an insufficiently developed system for laboratory diagnosis of hemostasis disorders in most regions of our country. The number of patients with hemophilia in Russia is just over 7.5 thousand people, and with von Willebrand disease there should be about 16 thousand. The exact figure has not been established. Rare hereditary forms of coagulopathies - hypoproconvertinemia (factor VII deficiency), hypo- and afibrinogenemia, deficiency of factors XII, XIII, XI, V are extremely rare.
Hereditary coagulopathies are incurable, but the possibilities of modern drug therapy make it possible to provide patients with a duration and quality of life comparable to that in the general population.
Hemophilia, von Willebrand disease and hypoproconvertinemia belong to the group of social diseases, and without government support, people suffering from hereditary forms of coagulopathies are doomed. This is primarily due to the provision of expensive drugs for blood clotting factors, which patients receive for life at home. Treatment of these patients throughout the world is carried out in specialized hemophilia centers that maintain a medical register of patients with hereditary coagulopathies, and is based on national standards and treatment protocols. The medical register allows you to track the dynamics of a patient’s health over many years and adjust treatment.
"Calling card" of hemophilia
The clinical course of hemophilia and von Willebrand disease is different, but may have some similarities. The presence of hemarthrosis is a “calling card” of hemophilia, but it should be remembered that hemarthrosis can also be observed in von Willebrand disease (the so-called type III), and sometimes with severe hypoproconvertinemia (factor VII deficiency). Hemophilia is a disease caused by various types of mutations in the factor 8 or 9 gene, which results in a hereditary deficiency of clotting factor VIII (hemophilia A) or IX (hemophilia B).
These genes are localized on the long arm of the X chromosome and are inherited in a recessive manner, transmitted through women only to male children. In the population, the level of factors VIII and IX varies from 100 ± 50%, but in women - conductors of hemophilia - it may be lower than normal due to the functional inferiority of one of the two genes. The severity of hemophilia depends on the level of factor VIII or IX activity. In severe hemophilia, factor VIII (or IX) is absent or shows residual activity (less than 2%). In this case, the disease usually appears from early childhood.
Bleeding is typical when the integrity of the mucous membranes and skin, hematomas (bleeding from the umbilical cord, cephalohematoma, ecchymosis) are violated. When the child begins to walk, the first hemorrhages in the joints appear. In the moderate form (from 2 to 5%), damage to the musculoskeletal system is also noted, and in the mild form (more than 5%), the disease usually manifests itself during injuries and surgical operations, which may be accompanied by severe bleeding due to the rapid depletion of endogenous factor VIII or IX . Late diagnosis of the disease can lead to tragic consequences.
Today such a scenario can be avoided
The occurrence of primary hemarthrosis, even after its visible elimination, causes invisible changes in the cartilage tissue of the joint, which can be recorded on an NMR tomogram. Disintegrated red blood cells form an environment for the occurrence of a secondary aseptic inflammatory process, and the resulting hemosiderin is deposited in the cartilage tissue of the articular surfaces.
Subsequent hemorrhages expand the affected area, lead to the development of chronic synovitis, contributing to the occurrence of “spontaneous” hemorrhages that accompany the patient throughout his life, ultimately leading to ankylosis and muscle atrophy. Before reaching adulthood, such patients previously became disabled with multiple lesions of the musculoskeletal system. During the life of these patients, as a rule, there are massive hematomas, gastrointestinal bleeding and other various hemorrhages that pose a threat to the patient’s life or lead to his death. Modern drug therapy makes it possible to avoid such a scenario for the development of the disease.
It should be noted that throughout the life of a hemophilia patient, the level of the factor does not change, so replacement therapy remains the only non-alternative solution today. The constant presence of factor VIII at least 5% creates conditions for normalizing the rate of clot growth, and clinically determines the absence of “spontaneous” hemorrhages.
It is known that the half-life of factor VIII and IX inactivation (T?) is short. On average, it is 12 and 24 hours, respectively, but it can vary significantly for each patient. In hemophilia A, this range for factor VIII ranges from 7 to 20 hours. This indicator is important to consider when correcting the hemostatic system in patients with hemophilia, especially if treatment is carried out for a long time.
Studies conducted in our center using the method of spatial dynamics of blood coagulation make it possible to assess the level of “sufficiency” of replacement therapy in patients with coagulopathies. Compensation for the level of the missing coagulation factor should be in the “corridor” between hypo- and hypercoagulation and maintained in a given range throughout the patient’s life. Such replacement therapy to prevent hemorrhagic episodes should be determined individually. This selection, with the simultaneous use of classical methods for determining blood coagulation factors, allows you to set optimal therapy parameters calculated for a specific patient and determine the amount of drug required for treatment.
The most vulnerable link in the treatment of these patients is determining the need for prescribing blood clotting factors. All existing blood clotting factors are administered intravenously, i.e. their bioavailability is 100%, however, the catabolism of these complex protein structures depends on many individual parameters of the entire hemostatic system.
Today, preventive replacement therapy for patients with hereditary coagulopathies is selected empirically, often guided only by the initial diagnosis and the visible clinical result, which is assessed very subjectively. The same applies to single therapeutic doses aimed at stopping a hemorrhagic episode. For widespread clinical practice, such tests seem difficult, at least for today.
Treatment on demand and according to the situation
In order to formalize the standards of treatment for patients with hemophilia, a Protocol for the management of patients with hemophilia was developed and approved in 2005. This event fundamentally changed the quality of life of patients.
Replacement therapy with blood clotting factors can be prescribed both for the purpose of preventing hemorrhages - the so-called. “preventive treatment”: for patients with hemophilia A - 25 IU/kg body weight 3 times a week; for patients with hemophilia B - 25 IU/kg - 2 times a week, and to stop bleeding (symptomatic hemostatic therapy - “treatment on demand” - from 20 to 50 IU/kg and further depending on the clinical situation). These two treatment models can be alternated throughout the patient's life and are the “cornerstone” of the protocol.
Treatment of the disease can be carried out not only for the purpose of preventing hemorrhages. The protocol for the management of patients with hemophilia provides a model for the treatment of existing bleeding or hemorrhage. In this case, the peak level of factor VIII or IX 30 minutes after injection should be 40-100%, depending on the clinical situation.
Injections of the drug are repeated every 12 hours for hemophilia A and every 24 hours for hemophilia B at a dose of ? from initial to disappearance of symptoms of hemorrhage. Our experience shows that this treatment regimen is acceptable for patients with isolated hemorrhagic complications. As with many other diseases, in hemophilia it is easier to prevent hemorrhages rather than treat their complications.
General recommendation document
Nevertheless, attending hematologists should remember that this document is of a general recommendation nature, and the approach to treating each patient should be individual, sometimes taking into account the psycho-emotional profile of the patient and even his family members.
Hereditary coagulopathies are incurable, but the possibilities of modern drug therapy make it possible to provide patients with a duration and quality of life comparable to that in the general population.
Intravenous administration of the drug is carried out by the patient himself or his relatives after completing a specialized training program, and the patient is monitored by a hemophilia center or a hematologist.
This treatment is called "home treatment" and is used throughout the world. In our country, all patients with hemophilia are currently undergoing “home treatment”.
It has also been established that the phenotype of the disease may differ from its genotype. Not all patients with the same level of factor VIII or IX require the same treatment. 10-15% of patients with severe hemophilia A do not need preventive treatment at all, i.e. in the constant administration of drugs, however, all patients with hemophilia, without exception, require lifelong provision of blood clotting factors VIII or IX. This also applies to other hereditary forms of coagulopathies - von Willebrand disease and hypoproconvertinemia (factor VII deficiency).
Modern antihemophilic drugs of blood coagulation factors VIII and IX, from the point of view of etiotropic therapy aimed at achieving a clinical result, do not differ significantly from each other. All of them compensate for the level of the missing clotting factor to the same extent. Their activity is expressed in standard international units (IU). Conventionally, they can be classified into blood coagulation factors VIII containing von Willebrand factor and those not containing. The quantity and quality of von Willebrand factor may vary significantly between them. There are drugs in which the content of von Willebrand factor is increased. There is an isolated von Willebrand factor that does not contain factor VIII.
Recombinant clotting factors
Factor VIII plasma preparations that do not contain von Willebrand factor, the amount of which is not changed in patients with hemophilia, include drugs that have undergone affinity chromatography, as well as genetically engineered (recombinant) blood coagulation factors VIII (INN: octocog alpha). The Octocog alpha group of drugs is represented by three generations of drugs (classification is arbitrary): the first, containing added albumin, necessary to stabilize the factor VIII molecule, the second, containing traces of human albumin, and the third, free from the presence of albumin.
The basis of effective therapy for hereditary coagulopathies is: early diagnosis, selection of the correct treatment model and complete, continuous provision of patients with blood coagulation factors VIII or IX. If the amount of the drug is not enough for treatment, the disease begins to progress steadily and can negate all previously made efforts.
There is a recombinant factor VIII with a modified molecule, i.e. removed glycoprotein fragment (moroctocog alpha). The experience of its use is not as extensive as that of previous drugs. For patients with hemophilia B, coagulation factor IX preparations can be used - recombinant (nanokog alpha) and obtained from donor plasma, as well as prothrombin complex preparations (PPSB).
In patients with von Willebrand disease, it is more appropriate to use drugs containing a physiological or greater physiological ratio of von Willebrand factor to factor VIII. These drugs were developed specifically for the treatment of patients with von Willebrand disease, and the contents of the latter are indicated on the bottle label.
Almost absolute virus safety
The accumulated experience in the use of recombinant and plasma drugs indicates almost absolute viral safety against the viruses HIV-1, HIV-2, hepatitis B and C. Over the past 20 years, not a single case of infection of a recipient has been reliably recorded, however, discussions based on the “potential threat" continues.
Complications in hemophilia, as a consequence of treatment, are also inevitable. The most dangerous, but rare in Russia (about 3-5% of patients with hemophilia A) is the emergence of resistance to therapy, caused by the formation of immunoglobulins, often class G, the so-called. autoantibodies that selectively block the procoagulant activity of the factor VIII molecule. In hemophilia B, the formation of antibodies is extremely rare (less than 1-2%).
Cases of the formation of antibodies to von Willebrand factor have been described. Antibodies to factor VII are practically not formed. It is important for practicing hematologists to know that antigenic stimulation (drug administration) can stimulate the immune response and cause complete tolerance to replacement therapy. An inhibitor should be suspected if previously effective therapy is ineffective. The etiology and pathogenesis of this phenomenon remain unclear.
Diagnostics in regions is not available
Therapy and its monitoring in patients with hemophilia complicated by an inhibitor poses significant difficulties even for experienced specialists and laboratory workers. Fundamentally, there are two types of therapy: activation of the hemostatic system through shunting (bypass) pathways. For this, recombinant activated factor VII or prothrombin complex (activated) is used - blood coagulation factors II, VII, IX, X in combination. The second treatment option is immune tolerance induction, usually the Bonn Protocol. In hospital practice, plasmapheresis or immunoadsorption with protein A can be used, but this therapy leads to a temporary decrease in the titer of the inhibitor and is currently rarely used. Treatment of inhibitory hemophilia patients represents a serious medical and social problem.
In von Willebrand disease, characterized by decreased activity of von Willebrand factor, ristocetin-induced platelet aggregation, often with decreased levels of factor VIII and factor VIII antigen, it is important to determine whether there is a deficiency in the production of von Willebrand factor (quantitative deficiency - type I) or the structure of the factor molecule synthesized defective (qualitative deficiency - type II).
Diagnosis of the disease is quite complex and often unavailable in the regions, because requires complex coagulation studies. Important symptoms of the disease are prolonged nosebleeds, in women, prolonged mensis, which can lead to iron deficiency anemia, prolonged bleeding after tooth extraction or minor surgical interventions, and the manifestation of the same symptoms in close relatives (father, mother, brother, sister). Patients with such symptoms should be examined in specialized centers.
Extremely rare disease
An extremely rare disease is hereditary factor VII deficiency. With a pronounced deficiency of this factor, severe hemorrhagic syndrome is observed, accompanied by hematomas, hemarthrosis, and in women - life-threatening menorrhagia.
To treat patients with hypoproconvertinemia, blood coagulation factor VII (plasma) can be used at a dose of 30-40 IU/kg every 8-10 hours. In its absence, activated eptacog alfa at a dose of 20-40 mcg/kg every 2-4 hours until bleeding stops completely.
The basis for effective therapy for hereditary coagulopathies is: early diagnosis, selection of the correct treatment model and complete, continuous provision of patients with blood coagulation factors VIII or IX. If the amount of the drug is not enough for treatment, the disease begins to progress steadily and can negate all previously made efforts.
Monitoring of patients with hereditary coagulopathies is carried out throughout their lives in specialized centers, often together with doctors of other specialties if concomitant diseases arise. The organization of such centers in Russia is the organization of “treatment technology” for patients suffering from disorders of the blood coagulation system.
Causes of poor blood clotting
Based on the above, we can name the main causes of coagulation disorders:
- Vascular wall defects are hereditary (associated with collagen abnormalities) and acquired (immune or inflammatory vascular lesions).
- Pathology of platelets. Platelet dysfunction sometimes causes significant bleeding. It includes thrombocytopenia (quantitative changes in platelets) and thrombocytopathies (changes in platelet quality). Acquired thrombocytopathies are caused by taking non-steroidal anti-inflammatory drugs, dipyridamole , antibiotics , uremia , heart valve pathology, and the use of extracorporeal circulation.
- The cause of a bleeding disorder may be a lack of factors (there are thirteen of them) of the blood coagulation system or a reduced synthesis of one or more factors. Prothrombin (factor II) is the main component of blood clotting. It is a precursor of thrombin and is involved in the formation of a clot (thrombus). Fibrinogen (factor I) is produced in the liver. In the coagulation cascade, it is converted into fibrin, which participates in clot formation. The deficiency of this factor was mentioned above.
- Deficiency of factor XI is associated with hemophilia C, factor VIII with hemophilia A , and factor IX with hemophilia B. Among hereditary coagulopathies, the most common (in 95% of cases) deficiency of factors VIII and IX. Deficiency of factors VII, X, V, XI is only 1.5%. Acquired deficiency of prothrombin complex factors (II, VII, X, V) occurs in liver diseases, jaundice , dysbacteriosis , as well as in overdose of vitamin K .
- The cause of bleeding can also be increased fibrinolysis , that is, excessive fibrinolytic activity. This may be a hereditary deficiency of alpha2-antiplasmin or increased formation of plasminogen activators and impaired excretion of these activators in liver diseases.
Symptoms
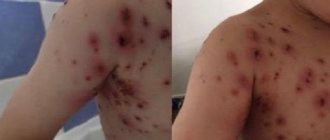
Whatever the cause of coagulopathy, the main symptom is bleeding of varying severity - from small bruises to severe bleeding during injuries (including minor ones). On the skin side, patients experience small petechiae , hematomas , bruises at injection sites, nasal and gingival bruises, heavy uterine bleeding in women, and often gastrointestinal bleeding.
Thrombocytopathies , both congenital and acquired, are not accompanied by severe hemorrhages . Bleeding in such patients can develop only during operations, injuries and tooth extraction. The most common manifestations are bruising and periodic nosebleeds and gum bleeding.
Thrombocytopenic purpura is a very common disease, especially among women 20-30 years old. Patients develop petechiae , bloody blisters that rise above the skin, bleeding gums and heavy uterine bleeding. The disease begins either gradually or acutely with hemorrhagic syndrome. According to its manifestations, there are two types of purpura: “wet”, when hemorrhages are combined with bleeding, and “dry”, if the patient has only skin hemorrhages. Hemorrhagic syndrome on the skin is observed in 100% of patients.
The number of hemorrhages can be single or multiple. Cutaneous hemorrhagic syndrome is characterized by:
- Various hemorrhagic rashes - petechiae and large hemorrhages.
- Inconsistency of hemorrhages with the degree of injury.
- Spontaneous appearance at night.
- Different colors of skin hemorrhages depending on the age.
- Painless.
- Asymmetry of elements.
- Hemorrhages in the soft palate and tonsils, sclera, fundus. Scleral hemorrhage sometimes precedes severe cerebral hemorrhage, which occurs quickly and progresses.
- Manifested by dizziness , headache , convulsions .
With von Willebrand disease, there is a tendency to intradermal hemorrhages, effusions of blood into the mucous membranes and severe bleeding after injury.
Clinical manifestations and diagnosis of vitamin K deficiency
In cases where the above etiological factors are not promptly addressed, vitamin K deficiency increases, which ultimately leads to the development of hemorrhagic syndrome in the child. As a rule, the clinical manifestation of VKDK is noted after 3–4 weeks. life of a child, most often this occurs at 1.5–2 months of age. At the same time, it is very important to remember that the maximum effectiveness of treatment measures for VKDK is achieved in cases where therapy begins with minimal hemorrhagic manifestations [11, 17]. In this regard, during an objective examination, it is necessary to pay attention to even the most minor hemorrhagic symptoms, which will become the basis for clarifying their causes. At this age, hemorrhagic syndrome is characterized by low specificity, and the maximum effectiveness of treatment is achieved only with an etiopathogenic approach, so searching for the cause becomes very important. Considering that hemorrhagic syndrome in infants can develop catastrophically quickly, it is necessary, without wasting time, to collect anamnesis and urgently (according to cito!
) perform a clinical blood test with the study of platelets and platelet indices, a coagulogram, a biochemical blood test, determine the blood group and Rh factor, and also conduct neurosonography due to the risk of intracranial hemorrhage (Fig. 1).
When collecting a family history, special attention should be paid to the presence of diseases in the child’s closest relatives that are accompanied by hemorrhagic syndrome (thrombocytopenia, thrombocytopathy, coagulopathy, thrombophilia). It is also very important to find out whether the child is prescribed medications that may cause hemorrhagic syndrome. In addition, in cases where the child is breastfed, it is necessary to clarify which medications are used by the mother. Anamnestic risk factors such as prematurity, long-term antibiotic therapy, and parenteral nutrition without vitamin supplementation must also be taken into account. Separately, it is necessary to clarify whether the child has hereditary metabolic disorders, congenital infections, malformations of the liver, gall bladder, intestines, as well as severe acquired diseases of the hepatobiliary and intestinal systems (Fig. 1).
When examining a child with hemorrhagic syndrome, regardless of the nature and severity of clinical manifestations, it is necessary to evaluate the level of consciousness, the color of the skin and visible mucous membranes, the color of feces and urine, the condition of the peripheral lymph nodes, liver, and spleen. In this case, the presence of icterus of the mucous membranes and skin with simultaneous lightening of stool and darkening of urine suggests damage to the hepatobiliary tract with the development of cholestasis. In these cases, first of all, it is necessary to think that the hemorrhagic syndrome is probably caused by acquired disorders of secondary hemostasis, since the synthesis of plasma factors of the coagulation system occurs in the liver.
A separate analysis requires a detailed description of the clinical manifestations of hemorrhagic syndrome. Thus, in the case of increased bleeding and hemorrhage, it is necessary not only to indicate the location (mucous membranes, umbilical wound, gastrointestinal tract, injection site, etc.), but also to note the time when the indicated symptoms first appeared, and also to clarify whether development was spontaneous or was a response to provoking factors. For hemorrhagic manifestations on the skin and/or mucous membranes, the localization, prevalence, a morphological description of the elements, severity, etc. should be indicated. It is very important to note when the hemorrhagic manifestations first appeared, what, in the parents’ opinion, could have caused them, and indicate their duration . Thus, often with a deficiency of vitamin K in the body, the first manifestation of VKDK is prolonged bleeding from the site of blood collection for clinical analysis or from the site where the vaccine was administered. Taking into account the fact that blood sampling for a hemogram usually precedes vaccination, bleeding detected during this process should be an absolute indication for studying the number of platelets, their indices and coagulogram. In this case, vaccination cannot be carried out until results are obtained that exclude hemostasis disorders. It should be especially noted that our data, which are consistent with the results of studies by other authors, indicate that underestimation of the early manifestations of hemorrhagic syndrome and the lack of adequate therapy at this stage lead to a significant increase in the frequency of severe complications due to the further development of intracranial hemorrhages [11 , 17–21].
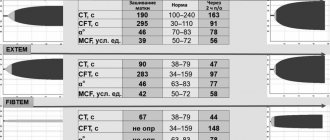
If thrombocytopenia is detected in a clinical blood test in the absence of abnormalities in the coagulogram, it is necessary to carry out differential diagnosis with a number of pathological conditions in which there is a decrease in the number of platelets (Fig. 2). If changes in the coagulogram are detected, and all laboratory indicators of primary hemostasis in a clinical blood test (platelet count, platelet indices, duration of bleeding) remain within normal limits, then we can conclude that coagulopathy occurs (Fig. 3). In this case, it is very important to differentiate at what stage of the cascade hemostasis system there is a failure. To do this, it is advisable to pay attention to the fact that in hemophilia there is a prolongation of the activated partial time (APTT) with normal indicators of the “external pathway” (prothrombin index (PTI), prothrombin time (PT), international normalized ratio (INR)) and the final stage of coagulation (thrombin time (TT), fibrinogen). Selective deficiency of coagulation factor VII is characterized by changes in the indicators of the “external pathway” (decrease in PTI, prolongation of PT and increase in INR) with normal values of the indicators of the “internal pathway” (APTT) and the final stage of coagulation (TT, fibrinogen). For a- or hypofibrinogenemia, typical changes in the coagulogram are prolongation of aPTT and TT, a decrease in PTI, an increase in PT and INR, and a decrease in fibrinogen levels [22].
In those cases when, in the absence of changes in the platelet link, disturbances are detected in the “internal pathway” (prolongation of APTT or complete absence of coagulation) and the “external pathway” (decrease in PTI, increase in PT, increase in INR or complete absence of coagulation), while As end-stage coagulation parameters (TB, fibrinogen) remain within normal limits, VCDC should be assumed (Fig. 4). These features of the coagulogram in VKDK are due to the fact that vitamin K-dependent blood coagulation factors are presented at different stages of the “internal pathway” and “external pathway” of coagulation, but do not take part in the final stage. Thus, coagulation factor IX is an obligatory component of the “intrinsic pathway”, so its deficiency will lead to a prolongation of the APTT or a complete absence of coagulation in case of severe deficiency. In turn, with insufficiency of coagulation factor VII, which is the key initiator of activation of the “external pathway,” there is a decrease in PTI, an increase in PT, an increase in INR, or a complete absence of coagulation when performing these tests, if there is a deep deficiency of factor VII. Coagulation factors II and X take part in both coagulation pathways; their deficiency will also be accompanied by changes in indicators characterizing both the “internal pathway” (prolongation of aPTT or complete absence of coagulation with deep deficiency) and the “external pathway” (decrease in PTI, increase in PT , increased INR or complete absence of coagulation). At the same time, in all these cases, the indicators of the final stage of coagulation (TV, fibrinogen) remain within normal limits, since vitamin K-dependent coagulation factors do not take part in coagulation at this level (Fig. 4).
Tests and diagnostics
Coagulological screening includes:
- prothrombin index;
- activated partial thromboplastin time;
- amount of fibrinogen;
- platelet count;
- bleeding time.
In case of isolated prolongation of activated partial thromboplastin time, proceed to the second stage of examination:
- do a correction test;
- activity of factors VIII, IX, XI, XII.
If the activity of factor VIII decreases, they proceed to the third stage of examination:
- lupus anticoagulant;
- specific inhibitor of factor VIII.
In children
Poor blood clotting in a child is often associated with immune thrombocytopenic purpura . Acute purpura develops between the ages of 2 and 9 years. This is an immune-related disease characterized by a constant (or periodic) decrease in platelets of less than 100 thousand. The disorder in children occurs 1-3 weeks after a viral infection. Such pathological reactivity can start not only under the influence of a viral infection, but also after taking medications, vaccination, exposure to temperatures (both low and high), surgical interventions or emotional stress. Against the background of normal health, the child develops a petechial rash (on the mucous membranes and skin), bruises, repeated nosebleeds and bleeding gums. In severe cases, there may be brain hemorrhages and stomach bleeding. Since antigens gradually leave the blood, in most people the disease goes away on its own after 2 months.
Vitamin K deficiency coagulopathy occurs in children in the first months of life and newborns . With Vit-K deficiency, the activity of several factors decreases: prothrombin , proconvertin , Christmas factor and Prower factor . hypocoagulation develops , accompanied by hemorrhagic syndrome.
Causes of Vit-K deficiency in a newborn:
- taking anticoagulants during pregnancy;
- antibiotics;
- anticonvulsants;
- severe damage to the pregnant woman’s liver and intestines;
- the presence of fetoplacental insufficiency;
- gestosis and preeclampsia .
The manifestations of this coagulopathy in newborns are not very specific - skin syndrome, increased bleeding during blood sampling and bleeding from the umbilical wound. With a lack of Vit-K, the duration of bleeding and platelet levels are within normal limits. Many authors recommend prophylactic administration of Vit K to all children immediately after birth - 2-3 administrations for the first 1.5 months, and in some cases weekly administration continues up to 3 months.
In later life, Vit-K deficiency coagulopathy is caused by breastfeeding only. At the same time, 78% of children develop massive intracranial hemorrhages. Much less frequently, the cause of decreased coagulability in children is disseminated intravascular coagulation syndrome in severe sepsis, congenital metabolic changes and hereditary coagulopathies.
Etiology of vitamin K-deficiency coagulopathy in children in the postneonatal period
VKDK is one of the leading causes of hemorrhagic syndrome in children in the first months of life [1–11]. It should be especially noted that VKDK, which develops in children in the postneonatal period, is still referred to as “late hemorrhagic disease of the newborn.” It is noted that this approach not only leads to terminological confusion, but also identifies diagnostic errors. Thus, earlier, in an anonymous survey of 348 pediatricians, we showed that the vast majority of respondents (2/3) associated late hemorrhagic disease of the newborn exclusively with the neonatal period and demonstrated a low level of awareness about the clinical manifestations, methods of diagnosis and treatment of VCD in infants [7]. The reason for these errors was the false idea among respondents that vitamin K deficiency has clear age intervals and is limited to the neonatal period. Moreover, in almost all cases, the pediatricians surveyed believed that late hemorrhagic disease of the newborn is due to the lack of prophylactic administration of vitamin K in the maternity hospital and is not associated with conditions such as exclusive breastfeeding, biliary atresia, severe damage to the hepatobiliary system or intestines, hereditary diseases accompanied by cholestasis [7].
Taking this into account, we proposed to exclude from circulation the concept of “late hemorrhagic disease of the newborn”, replacing it with the term “LCD” [7]. This approach will allow us to differentiate hemorrhagic syndromes that have the same pathogenesis (vitamin K deficiency and resulting coagulopathy), but differ in etiology and timing of manifestation. Thus, with hemorrhagic disease of the fetus and newborn (code P53 according to ICD-10), vitamin K deficiency develops in the perinatal period and is caused by insufficient transplacental intake. With this variant of VKDK, clinical manifestation is observed in the first 1–7 days of life. It should be especially emphasized that the administration of vitamin K to a child immediately after birth with high efficiency eliminates perinatal vitamin K deficiency and in the vast majority of cases prevents the development of hemorrhagic syndrome [1–5, 10–12]. In contrast, the development of VKDK in the postneonatal period, which is traditionally called late hemorrhagic disease of the newborn, is due to a number of reasons (exclusive breastfeeding, malformations or severe acquired lesions of the hepatobiliary system and/or intestines, hereditary diseases accompanied by cholestasis, long-term use of antibiotics and prolonged parenteral nutrition with insufficient vitamin K supplementation) and is not prevented by a single administration of vitamin K immediately after birth [1–5, 7–11, 13–17]. Taking this into account, it should be recognized as erroneous that the administration of vitamin K in the early neonatal period prevents the development of VKDK in the subsequent months of the child’s life. In this regard, when discussing this issue, it is very important to emphasize that vitamin K deficiency in the postneonatal period is due to completely different reasons. At the same time, their early detection allows for timely replacement therapy, which corrects vitamin K deficiency and prevents the development of VKDK.
Poor blood clotting during pregnancy
A normal pregnancy is always accompanied by important biochemical changes, including in the hemostatic . However, there are pathological conditions that lead to blood clotting disorders. Coagulopathy in pregnancy, what is it? This is a pathological condition that occurs with impaired coagulation and an increased risk of bleeding.
Pathological bleeding in pregnant women can be caused by:
- Congenital disorders in the coagulation system.
- Werlhof's disease.
- Moschkowitz disease.
- Antithrombin deficiency , which often occurs during pregnancy.
- Preeclampsia.
- Eclampsia.
- DIC syndrome associated with preeclampsia. At the first stage, intravascular coagulation occurs, and then the coagulation system is depleted.
- HELLP syndrome, which is also associated with preeclampsia (hemolysis, elevated liver enzymes and thrombocytopenia). Disseminated intravascular coagulation syndrome, a combination of thrombosis and bleeding.
- HELLP syndrome combines a triad of manifestations: hemolysis , decreased platelet levels and increased liver enzymes. During pregnancy with severe preeclampsia, the frequency of this syndrome reaches 20%.
- It develops during full-term pregnancy, premature birth and even after childbirth. HELLP syndrome is considered a subtype of preeclampsia. Diagnostic criteria for this syndrome: platelets less than 100×10 in 9/l, transaminases 2-3 times higher than normal, hemolysis of erythrocytes, bilirubin more than 20.5 µmol/l.
Women experience nausea , swelling and pain in the right hypochondrium. Pregnant women with HELLP syndrome are prescribed magnesium sulfate before and after birth for two days. Platelet transfusion is indicated when platelets are less than 20 × 10 in 9/L if a natural birth is expected and when platelets are less than 50 × 10 in 9/L if a cesarean section . Thromboconcentrate is administered before delivery. Corticosteroids increase platelet levels, so their use is reasonable. In the postpartum period, plasma exchange is used.
In DIC syndrome, a short phase of hypercoagulation is replaced by hypocoagulation. Such changes occur if a woman loses 15-20% of her blood volume. In case of bleeding, a platelet concentrate is infused.
In case of bleeding and if the prothrombin time and activated thromboplastin time are increased, an infusion of fresh frozen plasma is performed. If it is not possible to administer plasma, clotting factor concentrates are administered. Severe hypofibrinogenemia , which is not corrected by plasma transfusion, is then transfused with cryoprecipitate . For hyperfibrinolysis and bleeding, tranexamic acid . Hemolytic-uremic syndrome in women is accompanied by thrombocytopenia , kidney damage and microangiopathy . This condition most often develops after childbirth. Plasma exchange in this pathology is not very effective - the woman requires hemodialysis .
Treatment of vitamin K deficiency conditions
Timely identification of the cause of hemorrhagic syndrome makes it possible to prescribe adequate etiopathogenetic therapy in the early stages of the disease, which helps not only to stop the pathological process, but also to prevent the development of complications. In cases where we are talking about VKDK in children in the first months of life, it should be remembered that in 50–75% of cases VKDK leads to intracranial hemorrhages, accompanied by a high incidence of adverse outcomes, and in surviving children - serious complications [1–5 , 7–11, 13–21]. At the same time, most authors emphasize that intracranial hemorrhages due to vitamin K deficiency develop some time after the appearance of skin and/or mucous hemorrhages, and in some cases - against the background of ongoing bleeding from the site of blood sampling or injection, which was not noticed in a timely manner or remained underrated. In this regard, it is advisable to once again emphasize the need for an emergency search for causes even with a slight severity of hemorrhagic manifestations. In cases where, based on the analysis of clinical and anamnestic data and the results of laboratory examination, VKDK is verified, it is necessary to immediately begin replacement therapy. The drugs of choice in this case are the so-called prothrombin complexes - drugs that contain all vitamin K-dependent coagulation factors (II, VII, IX, X), as well as proteins C and S. After relief of the hemorrhagic syndrome, the planned administration of the vitamin is justified K. In cases where it is not possible to use prothrombin complex preparations, single-group fresh frozen plasma (10 ml/kg body weight) and vitamin K are administered. It should be remembered that if the effect of replacement therapy with prothrombin complex preparations or fresh frozen plasma occurs already in period of their administration, then the only synthetic analogue of vitamin K registered in our country (menadione sodium bisulfite) will manifest its hemostatic effect only after 18–24 hours. In this regard, with the development of hemorrhagic syndrome caused by VKDK, one cannot limit oneself only to the administration of menadione sodium bisulfite - simultaneous use of drugs containing vitamin K-dependent factors (or prothrombin complex, or fresh frozen plasma) is required.
Prevention
It is impossible to influence congenital pathologies of the coagulation system, but people with such pathologies can take a more careful approach to their health, choice of profession and physical activity. Secondary prevention includes the following:
- Avoid physical activity associated with the possibility of getting bruised (sports, football, wrestling, figure skating, etc.).
- Oral hygiene that helps reduce the need for dental procedures and surgeries.
- Normalize weight, which puts stress on the joints and increases the risk of bleeding into the joint cavity.
- Avoid the use of medications that affect clotting. Among such drugs are acetylsalicylic acid , Clopidogrel , Caffeine , Ibuprofen , Naproxen , nitrofurans , barbiturates , carbenicillin .
- Women with congenital factor deficiencies should consult and be examined by a geneticist and find out the risks of having a child with a congenital blood clotting disorder.
Similarly, there is no primary prevention of thrombocytopenic purpura , and secondary prevention is limited to preventing exacerbations. Patients should not be exposed to the sun; they are contraindicated from working in conditions of elevated temperature (hot shop, food shop near the stove). Children are exempt from physical education. After each case of ARVI, it is mandatory to test the blood.
Consequences and complications
With coagulopathies, the following complications may occur with varying probability and severity:
- Anemia.
- Hematuria (blood in the urine).
- Heavy and prolonged periods.
- Brain hemorrhage
- Gastrointestinal bleeding.
- Hemorrhage into the structures of the eye, pleurisy, compression of the larynx and trachea by hematomas.
- Hemorrhage into the joints and, as a consequence, the development of arthritis , arthrosis , osteoporosis and posthemorrhagic bursitis .
- Intravertebral hemorrhages.
- In rare cases, death due to massive bleeding.
Forecast
Acute forms of idiopathic purpura disappear within a few months and it often happens that the disease does not recur. In the chronic form, it is possible to induce remission, but the disease often recurs.
When using modern medications and following all recommendations for lifestyle changes and employment, thrombocytopenic purpura has a favorable prognosis.
Untreated hemophilia Lack of treatment often leads to joint pathology ( hemophilic arthropathy ), which then requires the use of crutches and wheelchairs or special orthopedic treatment. During treatment, life expectancy is practically no different from healthy individuals.
List of sources
- Fatkullin I.F., Zubairov D.M. Hereditary and acquired defects of the hemostatic system in obstetric and gynecological practice. M., 2002. P. 64.
- Galstyan G.M., Sukhanova G.A. Introduction to hemostasis, modern blood products and their effect on coagulation // Medical Council. 2013. from 11-13.
- Barkagan Z.S. Diagnosis and controlled therapy of hemostasis disorders / Z.S. Barkagan, A.P. Momot. – M.: Newdiamed, 2001. – 296 p.
- Barinov S.V., Dolgikh V.T., Medyannikova I.V. Hemocoagulation disorders in pregnant women with gestosis. Journal of Obstetrics and Women's Diseases. – 2013; 62 (6): 5–12.
- Degtyarev D.N., Karpova A.L., Mebelova I.I. and others. Draft clinical recommendations for the diagnosis and treatment of hemorrhagic disease of newborns // Neonatology. 2015. No. 2. P. 75–86.
Amniotic fluid embolism
This complex disorder is characterized by the sudden onset of hypotension, hypoxia, and consumption coagulopathy and has a variable clinical presentation. During delivery or immediately after birth, the mother develops shortness of breath, convulsions, respiratory and cardiac arrest, which is complicated by disseminated intravascular coagulation, massive bleeding and ends in death.
Pathogenesis. Amniotic fluid enters the microcirculation due to a violation of the physiological barrier that normally exists between the maternal and fetal vascular systems. The gateway for elements of fetal tissue to enter the maternal vascular system can be trauma, amniocentesis, but most often it is rupture of the cervix and lower uterine segment during childbirth. Caesarean section also provides the opportunity for mixing of maternal blood and fetal tissue.
The pathophysiological cascade is triggered by numerous chemokines and cytokines. After a short initial phase of pulmonary and general hypertension, a decrease in peripheral vascular resistance and stroke working volume of the left ventricle occurs. Transient but profound oxygen desaturation in the initial phase leads to neurological complications in those patients who survive. Those who survive the initial cardiovascular collapse develop a secondary phase of pulmonary injury and consumptive coagulopathy.
Uterine hypertonicity is not the cause of embolism, as is often believed. Uterine blood flow completely stops when intrauterine pressure increases to 35-40 mm Hg. Art. So, hypertensive uterine contractions are the least likely time for fetomaternal exchange. According to modern research, the use of oxytocin is not associated with an increase in cases of amniotic fluid embolism.
Diagnosis of amniotic fluid embolism was previously based on the presence of fetal squamous epithelial cells in the central pulmonary vessels of the mother, which was considered a pathognomonic sign of embolism. Modern research shows that fetal flat cells, trophoblast and other products of fetal origin can be found in the maternal vascular system without connection with amniotic fluid embolism. In summary, the presence of fetal tissue in the pulmonary vessels is neither a sensitive nor a specific diagnostic test. The diagnosis of amniotic fluid embolism is determined by excluding other causes of death.
Treatment of amniotic fluid embolism consists of stabilizing vital functions (pulmonary-cardiac resuscitation), restoring blood volume, and treating coagulation disorders. But there is no reliable data that any intervention improves the prognosis for amniotic fluid embolism. If amniotic fluid embolism occurs before delivery, an urgent cesarean section is recommended in order to save the life of the fetus. But if the mother’s hemodynamics are unstable, the possibility of such intervention becomes more difficult.
If the patient survives, the prognosis for the mother and fetus is complicated by the possibility of serious post-hypoxic neurological disorders. The consequences for the fetus worsen as the interval from the embolic episode to delivery increases.