1.What is von Willebrand-Jurgens syndrome?
Von Willebrand-Jurgens syndrome
(constitutional thrombopathy) is the most common congenital bleeding disorder. The disease is also often called "hereditary antihemophilia". The picture of the disease is indeed very similar to hemophilia. When wounded, bleeding continues longer than usual. Hematomas form subcutaneously.
It is known that platelets are involved in the formation of a blood clot that closes a wound. However, without the participation of a specific protein (von Willebrand factor)
coagulation is impaired due to the inability of platelets to adhere to the walls of blood vessels. In addition to this, the disease causes vascular permeability and fragility. In patients with von Willebrand-Jurgens syndrome, the homeostasis chain is disrupted, so the risk of any bleeding is very high. Sick women are especially at risk because their periods are protracted, even to the point of exhaustion. At the same time, there are cases where pregnant patients with von Willebrand-Jurgens syndrome successfully carry a child, and childbirth occurs without complications.
It has also been noted that the course of the disease changes with age. In some cases, a complete cure occurs or the degree of impairment is noticeably reduced.
A must read! Help with treatment and hospitalization!
Causes
One of the parts of the hemostasis system of the human body is von Willebrand factor (VWF), which performs two main functions:
- triggers the mechanism of adhesion (gluing) of platelets to the site of damage to the blood vessel;
- stabilizes coagulation factor VIII circulating in the blood.
Various genetic disorders cause a defect in the synthesis of von Willebrand factor, as a result of which it is produced in insufficient quantities (in some cases, its synthesis is impossible at all). A variant of the disease is also possible in which the amount of VWF is optimal, but the protein itself is defective and cannot perform its functions. As a result, VW deficiency suffers, according to various sources, from 0.1 to 1% of the population. However, often this disease is mild and may not be diagnosed at all.
2.Types of disease depending on the severity of the disease
The severity of the disease is classified by type:
- 1 type
The most common and easiest form. The decrease in blood clotting is insignificant. The patient may not be aware of the presence of the syndrome. Von Willebrand-Jurgens factor is present in the blood, but in insufficient concentration. - Type 2
There is von Willebrand-Jurgens factor in the blood, but it does not work. The form is rarer, but also more dangerous. It is inherited in some cases - with a certain combination of genetic characteristics of the parents. It manifests itself as minor bleeding without visible prerequisites. - Type 3
Only 5% of patients have von Willebrand-Jurgens factor completely absent. Serious risk of bleeding, anemia and complications during operations.
Most often, von Willebrand-Jurgens syndrome is a congenital pathology. The disease is acquired against the background of some other conditions and disorders.
Since von Willebrand-Jurgens syndrome is based on a clotting disorder, blood can appear where bleeding should not occur in a healthy person. The most commonly observed:
- nosebleeds;
- menstrual bleeding in women;
- blood in urine;
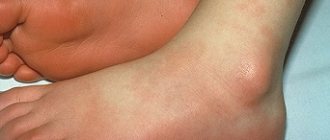
- subcutaneous hematomas;
- bleeding of the oral mucosa;
- dark stools (stained with blood);
- joint hemorrhages as a result of movement.
Even with type 1 of the disease, the patient, when visiting any doctor, must inform him of his diagnosis
, since many manipulations, studies and procedures involve microtraumas that are not dangerous for healthy people. The patient's condition must be taken into account when prescribing and choosing a particular treatment regimen for him.
Visit our Therapy page
Inherited illness: why is von Willebrand disease dangerous?
Today, according to official statistics, about 8.5 thousand patients suffer from hemophilia in Russia, although this pathology was once considered a royal disease. Hemophilia is a hereditary disease that usually affects only men, while women act as carriers of the gene. Of course, the most famous carrier of hemophilia in history was Queen Victoria. Presumably, this mutation occurred in her de novo genotype, since there were no hemophilia patients in her parents' families. The most common forms of the disease are classical hemophilia type A and hemophilia type B, in which a deficiency of clot-forming factor VIII or plasma factor IX (Christmas) develops.
Unlike hemophilia, which affects only men, there is another hereditary hemorrhagic diathesis that also affects women - von Willebrand disease. This pathology, inherited in an autosomal dominant manner, is much more difficult to diagnose and often the patient learns about it only during severe injuries, surgical interventions and other critical conditions. In the world, von Willebrand disease occurs in 1 person out of 100. In Russia, according to official data, the prevalence is 0.001%, but many experts believe that the real number of patients is much higher. The low detection rate of the disease can be explained by the predominance of mild and asymptomatic forms, as well as the complexity of diagnosis. The majority of patients with hemophilia and von Willebrand disease are people of working age, which is why measures aimed at preventing complications come to the fore. In von Willebrand disease, there is a deficiency of von Willebrand factor, a protein that plays an important role in regulating platelet adhesion to subendothelial collagen and protects factor VIII from proteolysis. In addition, the serotonin content decreases and pathological dilatation of blood vessels and an increase in their permeability develop. It is worth emphasizing that it is with this pathology that the longest bleeding is observed, since all three parts of hemostasis are damaged. There is also acquired von Willebrand syndrome, characteristic of patients with autoimmune and lymphoproliferative diseases and caused by the formation of an inhibitor against von Willebrand factor.
The clinical course of von Willebrand disease can significantly reduce the quality of life, lead to early and persistent disability, and in severe cases even death. For a long time, the disease was considered as a form of hemophilia, since many manifestations of von Willebrand disease are similar to the symptoms of this nosology. However, treatment for hemophilia has not always been effective in cases of von Willebrand disease. In this regard, in March 2015, a new List of Medicines came into force, in which the treatment of von Willebrand disease was included in a separate item.
The most pathognomonic symptoms of von Willebrand disease include bleeding from the mucous membranes of the mouth, nose, internal organs, as well as the appearance of long-lasting bruises. Unlike hemophilia, which is diagnosed at birth, von Willebrand disease, especially its mild forms, is diagnosed only in adolescence or during invasive procedures, when bleeding can be difficult to stop. The severity of manifestations can vary from mild forms to very severe variants with prolonged bleeding of a wide variety of localizations, the formation of hematomas and hemorrhages in soft tissues, joints and internal organs. With such forms, children became disabled by the age of 12-15 and rarely lived to be 30 years old.
Today, thanks to the achievements of world science, patients with bleeding disorders can live a full life, get an education, work, and successfully socialize. The duration and quality of their life is at the same level as that of a healthy person. If von Willebrand disease is suspected, a blood test is necessary for the qualitative and quantitative characteristics of von Willebrand factor. The most commonly used diagnostic tests are ristomycin cofactor activity, ristomycin platelet aggregation, collagen von Willebrand factor binding activity, factor VIII von Willebrand factor binding activity, or multimer assay. However, there are still several issues that need to be addressed. How often should diagnostics be carried out? Which patients need it? How to evaluate the norm? Moreover, the activity of von Willebrand factor is constantly changing. It can increase with stress or decrease with decreasing estrogen concentrations in women.
Treatment of von Willebrand disease is divided into specific and nonspecific. The most important pathogenetically determined point is the administration of hemotherapy containing a complex of factor VIII and von Willebrand factor. The question remains open: what bleeding should be treated with replacement therapy and what should be the dose of drugs with von Willebrand factor? Transfusion should begin several days before the proposed surgical operations, since correction of hemostasis occurs gradually. Transfusion of platelets or the use of drugs that affect platelets are not effective, since in this case platelet dysfunction occurs secondary. For mild to moderate severity and the development of microcirculatory type bleeding, the effectiveness of aminocaproic acid has been proven.
Currently in Russia there is a program “7 nosologies”, which is intended for the treatment of patients with seven rare and expensive nosologies at the expense of the federal budget. Thanks to this program, the percentage of disability has significantly decreased, the quality of life of patients has improved and the risk of life-threatening bleeding has decreased. The advantage of the “7 nosologies” program was the provision of medicinal care according to nosological principle, and not according to disability status. It is extremely important to maintain the current system of drug supply and centralized procurement of drugs, since for some categories of patients this program becomes the only opportunity to support and preserve their own lives.
Bibliography:
- Christine A. Lee, Rezan A. Kadir, Peter A. Kouides: Inherited Bleeding Disorders in Women.
- Chandler WL, Peerschke EI, Castellone DD, Meijer P (June 2011). “Von Willebrand factor assay proficiency testing. The North American Specialized Coagulation Laboratory Association experience." Am. J. Clin. Pathol. 135(6):862–9.
- "Molecular basis of von Willebrand disease and its clinical implications." Haematologica 89 (9): 1036. 1 September 2004.
Prepared by:
Drapkina Yu.S.
4. Treatment of von Willebrand-Jurgens syndrome
Specific treatment
this disease does not exist. Medical care is usually aimed at stopping bleeding that occurs. If surgical treatment is necessary, measures are taken to prevent bleeding (plasma preparations with a high content of von Willebrand factor, desmopressin, aminocaproic acid are used). In cases of severe forms of the disease, when there is a high risk of bleeding even from household injuries and bruises, medications are prescribed that prevent the destruction of blood clots after they form. As an emergency, fibrin glue or prombin powder is used to help stop bleeding from the wound.
Patients should
know and avoid taking medications whose side effect is a decrease in blood clotting (heparin, aspirin, ibuprofen, naproxen, medications containing salicylates). Patients with severe forms should be able to inject themselves with clotting factors. It is also necessary to know and identify the signs of hidden bleeding. To reduce household injuries, you need to monitor your weight, maintain good physical shape, while avoiding dangerous sports.
Can BV be cured?
It is impossible to completely cure von Willebrand disease, just as it is impossible to eliminate the gene defect that causes it. People with BV are prescribed therapy to prevent bleeding, especially postoperative and postpartum bleeding. These are drugs that increase the level of vWF in the blood - desmopressin, antihemophilic factor and others.
Women suffering from heavy periods and menorrhagia are prescribed oral contraceptives.
Regarding the delivery, interpretation of tests and treatment of von Willebrand disease, you can contact the hemostasiologists of the Medical Women's Center.
Symptoms
The symptoms of von Willebrand disease are extremely diverse: from minor episodic bleeding to massive, debilitating bleeding, leading to severe blood loss.
Signs characteristic of von Willebrand disease:
- severe, prolonged or spontaneously renewing bleeding after minor surgical interventions, tooth extraction;
- subcutaneous hematomas that appear after minor traumatic impacts or spontaneously
- bleeding for more than 15 minutes after minor injuries or spontaneously recurrent bleeding 7 days or more after injury;
- hemorrhagic skin rash;
- severe anemia;
- intense, prolonged menstruation;
- spontaneous nosebleeds lasting more than 10 minutes or requiring medical intervention due to the intensity;
- blood in feces in the absence of gastrointestinal pathology that can provoke the development of gastrointestinal bleeding.
More often, increased bleeding is observed in childhood, decreasing as they grow older, and subsequently there is an alternation of exacerbations and remissions.
Diagnostics
Diagnosis of von Willebrand disease requires the indispensable participation of hematologists. It is impossible to make a diagnosis at the level of a clinic or children's consultation due to the lack of capabilities of the laboratories of these institutions to conduct specific diagnostic tests and enzyme-linked immunosorbent technologies. The doctor assumes the disease when interviewing relatives, examining the patient and taking into account information from the anamnesis.
Moreover, available tests vary in sensitivity and diagnostic value. Therefore, an algorithm for examining the suspected patient has been developed.
- First, a coagulogram is studied with all indicators of coagulation, including bleeding time. The test can be done at your clinic. If pathological abnormalities are detected, the patient is referred to the hematology center.
- To identify a specific type of disease and the qualitative side of the defect, a comparison of the impaired aggregation ability under the influence of ristocetin with the normal one is used - under the influence of collagen, thrombin, ADP, and adrenaline.
- The main method of detecting a decrease in the amount of factor VIII in the blood is to determine the activity of the patient’s platelets treated with formaldehyde in reaction with a solution of ristocetin.
- Using the collagen-binding technique, the impaired functional ability of factor VIII and the specific type of disease are identified.
In the diagnosis of concomitant lesions of the mucous membranes it is necessary:
- examination by an otolaryngologist;
- esophagogastroduodenoscopic examination;
- colonoscopy (examination of the intestines).
For treatment, it is important to identify vascular formations in the form of tortuosity, angiomas, extensions up to 2 mm, which contribute to bleeding.