Общие сведения
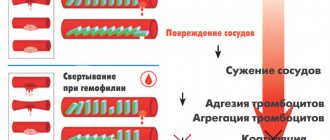
Система свертывания крови — это сложный последовательный каскад реакций, происходящих в организме, который направлен на остановку кровотечения. Процесс свёртывания — важная защитная реакция организма, благодаря которой поддерживается постоянный объём циркулирующей крови. В системе гомеостаза участвуют множество компонентов, основные из которых указаны на рисунке ниже.
Факторы свертывания находятся в крови в неактивном состоянии. Если кровеносный сосуд травмируется, запускается процесс свертывания, и все факторы последовательно активируются и обеспечивают формирование сгустка. Сам непосредственный процесс свертывания связан с превращением белка фибриногена (фактор I) в нерастворимый фибрин.
Коагулопатией называется болезнь, вернее группа заболеваний или состояний, в основе которых лежит нарушение свертываемости крови. Клиническим признаком коагулопатии является кровотечение. Приобретенные коагулопатии — самые часто встречаемые синдромы. При этом плохая свертываемость крови может быть обусловлена патологией разных звеньев системы свертывания: фибрин, тромбоциты или факторы свертывания крови. Если какое-либо звено этой системы не функционирует или отсутствует, то у человека будет продолжительное кровотечение и разовьется критическое состояния. Среди коагулопатий есть врожденные состояния и заболевания (гипофибриногенемия (дефицит I фактора), афибриногенемия, гемофилия А, болезнь Виллебранда, гемофилия В) так и приобретенные синдромы, возникающие при различных септических состояниях и заболеваниях почек и печени.
Гипофибриногенемия
В поздние сроки беременности уровень фибриногена равен 3-6 г / л. При активации коагуляции этот высокий уровень фибриногена может иногда играть защитную роль против клинически значимой гипофибриногенемии. Для обеспечения клинической коагуляции уровень фибриногена не должен быть меньше 1,5 г / л. При наличии значительной гипофибриногенемии кровяной сгусток цельной крови в стеклянной пробирке сначала может быть мягким, хотя его объем существенно не уменьшается. Но через некоторое время (около 30 мин) он становится маленьким, когда эритроциты и жидкость высвобождаются из него.
Фибрин и дериваты фибриногена. Продукты деградации фибриногена в сыворотке крови является чувствительным маркером коагулопатии, и их определение лежит в основе многих тест-систем (моноклональные антитела). Повышение содержания продуктов деградации фибриногена свидетельствует о выраженной коагулопатии потребления.
Патогенез
В патогенезе геморрагического синдрома присутствует:
- повреждение клеток эндотелия сосудов медиаторами воспаления или эндотоксином;
- активация протеина С, который ингибирует ФV и ФVIII и подавляет синтез ингибитора активатора плазминогена (последний способствует переходу плазминогена в плазмин, который расщепляет фибрин тромба);
- активация фибринолиза, которая играет роль в развитии не останавливающихся кровотечений и истощение ф I, II, V, VIII, ХIII;
- накопление в крови метаболитов, обладающих антикоагулянтным действием.
В патогенезе уремической тромбоцитопатии имеет значение недостаточность тромбоцитов, которая связана с действием токсических метаболитов плазмы. Кроме того, больным с уремией выполняют процедуру экстракорпорального кровообращения, при которой возникает дисфункция тромбоцитов из-за взаимодействия их с трубками и мембранами аппарата. При этом тромбоциты активируются и освобождают гранулы (дегрануляция тромбоцитов). Тромбоцитарная дисфункция вызывает настолько выраженное кровотечение, что требуется переливание тромбоцитарной массы.
Лекарственная тромбоцитопения связана с взаимодействием препарата (или его метаболита) и гликопротеина мембраны тромбоцита. В результате такого взаимодействия образуется иммуногенный комплекс — гликопротеин-препарат. Измененные тромбоциты удаляются из кровотока клетками РЭС. При лекарственной тромбоцитопении повышается уровень IgG и связанные тромбоцитами антитела к препарату. В основе идиопатической пурпуры — выработка антител против антигенов вируса. Тромбоциты повреждаются, адсорбируя на своей мембране вирусный антиген или же иммунный комплекс вирус-антитело.
Классификация
Коагулопатии делятся на наследственные и приобретенные.
- Наследственные формы связаны с генетически обусловленными изменениями стенок сосудов, аномалиями тромбоцитов и плазменных факторов крови. К наследственным коагулопатиям относится гемофилия А, болезнь Виллебранда, гемофилия В, дефицит различных факторов свертываемости.
- Приобретенные формы чаще всего связаны с поражением сосудов различной этиологии (иммунной, токсикоинфекционной и дисметаболической), поражением тромбоцитов, патологией факторов свертывающей системы и комбинацией всех этих факторов.
Различают следующие виды приобретенной коагулопатии:
- Нарушения тромбоцитарного гемостаза. Сюда относятся тромбоцитопении различного происхождения (связанная со снижением продукции тромбоцитов, связанная с повышенным их разрушением, обусловленная не иммунными причинами и иммунными — идиопатическая тромбоцитопеническая пурпура), HELLP- и гемолитико-уремический синдром, тромботическая тромбоцитопеническая пурпура, тромбоцитопатии. При тромбоцитопении может первично поражаться тромбоцитарный росток, тромбоциты могут перераспределяться и скапливаться в селезенке, а в крови их будет недостаточное количество. Также тромбоциты могут в большом количестве разрушаться (при красной волчанке и тромбоцитопенической пурпуре). Кроме тоготромбоциты могут потребляться в большом количестве при образовании тромбов (например, при ДВС- синдроме). Тромбоцитопатии характеризуются выработкой аномальных тромбоцитов, функция которых нарушена. Примером тромбоцитопатии служит болезнь Виллебранда и тромбастения Гланцманна.
- Виды коагуляционных нарушений. В эту группу входят нарушения гемостаза, связанные с передозировкой антикоагулянтов (гепарина, варфарина), гемодилюционная коагулопатия, витамин К-зависимые (при нарушении функции печени, плохом всасывании витамина К, приеме некоторых медикаментов).
- Коагуляционно-тромбоцитарные (смешенные) нарушения, развившиеся на фоне недостаточности печени и почек (печеночная и уремическая коагулопатия).
- Отдельно выделяется ДВС-синдром.
В системе свертывания крови важную роль играет фибриноген (фактор I). В норме его содержание 2-4 г/л. Часто у больных определяется гипофибриногенемия — снижение уровня фибриногена. Это состояние может быть наследственным, но чаще приобретенным, по причине недостаточного образования этого белка печени при ее поражении или повышенного растворения его (фибринолиз). При этом состоянии свертывание крови замедляется и в результате образуется рыхлый распадающийся сгусток. Проявляется гипофибриногенемия образованием синяков при незначительной травме, различными кровотечениями.
Однако у большинства пациентов сниженный уровень фибриногена никак не проявляется. Снижение уровня этого белка отмечается при циррозе, некрозе печени, метастазах в костный мозг, лейкозах, шоке, анемии, эклампсии, преждевременной отслойке плаценты, осложненных родах, сепсисе. Причиной острой гипофибриногенемии является внутрисосудистое свертывание крови, когда фибриноген усиленно потребляется. Многим пациентам со сниженным уровнем фибриногена лечение не требуется. Обильные менструации у женщин предотвращаются гормональными средствами и антифибринолитическими препаратами.
Афибриногенемия — полное отсутствие фибриногена в крови при нормальном уровне тромбоцитов. Это генетически обусловленное заболевание, но встречается очень редко. При этом состоянии любая травма приводит к кровотечению, гематомам, кровоизлияниям в суставы. Стоматологические манипуляции и операции сопровождаются значительной кровопотерей. Дети редко доживают до взрослого возраста. В лечении этой патологии применяется заместительная терапия: введение концентрата фибриногена, криопреципитата и свежезамороженной плазмы.
Наследственные коагулопатии: гемофилия и болезнь Виллебранда
Система свертывания крови человека представляет собой многокомпонентный и чрезвычайно сложный механизм, играющий важнейшую роль в защите всего организма.
Указанный механизм представлен тремя звеньями: сосудистым, тромбоцитарным и плазменно-коагуляционным. Заболевания, связанные с неспособностью факторов свертывания крови обеспечивать нормальный процесс образования сгустка, составляют обширную группу коагулопатий. Эта группа болезней представлена наследственными коагулопатиями и множеством приобретенных форм коагулопатий, которые являются следствием других заболеваний: цирроз и рак печени, аутоиммунные (системная красная волчанка, иммунная тромбоцитопеническая пурпура, геморрагический васкулит и т.п.), инфекционные заболевания, токсическое воздействие лекарственных средств и ядов, наследственные заболевания обмена веществ.
Наследственные заболевания гемостаза
Гемофилия и болезнь Виллебранда являются наиболее частыми наследственными заболеваниями системы плазменного звена гемостаза. Фактор Виллебранда и фактор VIII в плазме крови представлены в виде молекулярного комплекса, при этом фактор Виллебранда выполняет защитную роль для фактора VIII, оберегая его от разрушения протеином С.
Именно поэтому при отсутствии фактора Виллебранда уровень фактора VIII может быть значительно снижен. Таким образом, несмотря на различие данных заболеваний, они проявляют определенный молекулярно-биологический синергизм. В этой связи для практикующих врачей целесообразно рассмотреть два заболевания, занимающих более 90% наследственных коагулопатий, обусловленных дефицитом или функциональной несостоятельностью пламенных факторов свертывания крови — VIII, IX (гемофилия А или В соответственно), или фактора Виллебранда (болезнь Виллебранда).
Крайне редко встречается и дефицит фактора VII. Гипопроконвертинемия проявляется геморрагическим синдромом в зависимости от выраженности дефицита фактора VII. В тяжелых случаях отмечаются гемартрозы, гематомы, у женщин меноррагии.
И встречаются, и диагностируются нечасто
В целом наследственные коагулопатии встречаются нечасто, а диагностируются еще реже. На это есть две основные причины: низкая распространенность, часто сопряженная с субклиническим течением заболевания (при болезни Виллебранда) и недостаточно развитая система лабораторной диагностики нарушений гемостаза в большинстве регионов нашей страны. Количество больных гемофилией в России составляет чуть более 7,5 тыс. человек, а с болезнью Виллебранда должно быть около 16 тыс. Точная цифра не установлена. Редкие наследственные формы коагулопатий — гипопроконвертинемия (дефицит фактора VII), гипо- и афибриногенемия, дефицит факторов XII, XIII, XI, V встречается крайне редко.
Наследственные коагулопатии неизлечимы, но возможности современной препаратной терапии позволяют обеспечить больным продолжительность и качество жизни, сравнимые с таковой в общей популяции.
Гемофилия, болезнь Виллебранда и гипопроконвертинемия относятся к группе социальных заболеваний, и без поддержки государства люди, страдающие наследственными формами коагулопатий, обречены. Это связано в первую очередь с обеспечением дорогостоящими препаратами факторов свертывания крови, которые больные получают пожизненно в «домашних условиях». Лечение этих пациентов во всем мире проводится в специализированных центрах гемофилии, ведущих медицинский регистр больных с наследственными коагулопатиями, и базируется на национальных стандартах и протоколах лечения. Медицинский регистр позволяет отслеживать динамику здоровья пациента в течение многих лет и корригировать лечение.
«Визитная карточка» гемофилии
Клиническое течение гемофилии и болезни Виллебранда отличается, но может иметь некоторую схожесть. Наличие гемартрозов является «визитной карточкой» гемофилии, однако следует помнить, что гемартрозы могут отмечаться и при болезни Виллебранда (т.н. тип III), а иногда и при выраженной гипопроконвертинемии (дефицит фактора VII). Гемофилия — заболевание, обусловленное различными типами мутаций гена фактора 8 или 9, в результате которых возникает наследственный дефицит фактора свертывания крови VIII (гемофилия А) или IX (гемофилия В).
Эти гены локализованы на длинном плече X-хромосомы и наследуются по рецессивному признаку, передаваясь через женщин только детям мужского пола. В популяции уровень факторов VIII и IX варьирует от 100 ± 50%, но у женщин — кондукторов гемофилии— он может быть ниже нормы, вслед-ствие функциональной неполноценности одного из двух генов. Тяжесть гемофилии зависит от уровня активности фактора VIII или IX. При тяжелой форме гемофилии фактор VIII (или IX) отсутствует или проявляет остаточную активность (менее 2%). В этом случае заболевание обычно проявляется с раннего детства.
Характерны кровотечения при нарушении целостности слизистых и кожных покровов, гематомы (кровотечение из пуповины, кефалогематомы, экхимозы). Когда ребенок начинает ходить, появляются первые кровоизлияния в суставы. При средней форме (от 2 до 5%) также отмечается поражение опорно-двигательной системы, а при легкой (более 5%) заболевание обычно проявляется при травмах и хирургических операциях, которые могут сопровождаться сильными кровотечениями из-за быстрого истощения эндогенного фактора VIII или IX. Поздняя диагностика заболевания может привести к трагическим последствиям.
Сегодня такого сценария можно избежать
Возникновение первичного гемартроза даже после его видимого устранения вызывает невидимые изменения в хрящевой ткани сустава, которые можно зарегистрировать на ЯМР-томограмме. Распавшиеся эритроциты формируют среду для возникновения вторичного асептического воспалительного процесса, а образовавшийся гемосидерин откладывается в хрящевой ткани суставных поверхностей.
Последующие кровоизлияния расширяют зону поражения, приводят к развитию хронического синовита, способствуя возникновению «спонтанных» кровоизлияний, сопровождающих больного в течение всей его жизни, приводя в конечном счете к анкилозу и атрофии мышц. До совершеннолетия такие больные ранее становились инвалидами с множественным поражением опорно-двигательной системы. В течение жизни этих больных, как правило, отмечаются массивные гематомы, желудочно-кишечные кровотечения и другие всевозможные кровоизлияния, представляющие угрозу для жизни пациента или приводящие к его смерти. Современная препаратная терапия позволяет избежать такого сценария развития болезни.
Следует отметить, что в течение всей жизни больного гемофилией уровень фактора не меняется, поэтому заместительная терапия остается единственным на сегодняшний день безальтернативным решением. Постоянное присутствие фактора VIII не ниже 5% создает условия нормализации скорости роста сгуст-ка, а клинически обуславливает отсутствие «спонтанных» кровоизлияний.
Известно, что полупериод инактивации фактора VIII и IX (T?) невелик. В среднем он составляет соответственно 12 и 24 часа, однако у каждого пациента он может значительно варьировать. При гемофилии А такой разброс для фактора VIII составляет от 7 до 20 часов. Этот показатель важно учитывать при коррекции системы гемостаза у больных гемофилией, особенно, если лечение проводится в течение длительного времени.
Проводимые в нашем центре исследования с использованием метода пространной динамики свертывания крови позволяют оценить уровень «достаточности» заместительной терапии у больных с коагулопатиями. Компенсация уровня недостающего фактора свертывания крови должна находиться в «коридоре» между гипо- и гиперкоагуляцией и поддерживаться в заданном диапазоне в течение всей жизни пациента. Подобная заместительная терапия, профилактирующая геморрагические эпизоды, должна определяться индивидуально. Этот подбор с одновременным использованием классических методов определения факторов свертывания крови позволяет задавать оптимальные, рассчитанные для конкретного пациента, параметры терапии и определять необходимое для лечение количество препарата.
Самое уязвимое звено в лечении этих пациентов — определение потребности при назначении факторов свертывания крови. Все существующие факторы свертывания крови вводятся внутривенно, т.е. их биодоступность составляет 100%, однако катаболизм этих сложных белковых структур зависит от многих индивидуальных параметров всей системы гемостаза.
На сегодняшний день профилактическая заместительная терапия пациентам с наслед-ственными коагулопатиями подбирается эмпирически, чаще руководствуясь только первоначально поставленным диагнозом и видимым клиническим результатом, который оценивается весьма субъективно. То же самое касается и однократных лечебных доз, направленных на купирование геморрагического эпизода. Для широкой клинической практики подобные тесты представляются затруднительными, во всяком случае, на сегодняшний день.
Лечение по требованию и по ситуации
Для того, чтобы формализировать стандарты терапии больных гемофилией, в 2005 году был разработан и утвержден Протокол ведения больных гемофилией. Это событие принципиально изменило качество жизни пациентов.
Заместительная терапия факторами свертывания крови может назначаться как с целью профилактики кровоизлияний — т.н. «профилактическое лечение»: для больных гемофилией А — 25 МЕ/кг веса тела 3 раза в неделю; для больных гемофилией В — 25 МЕ/кг — 2 раза в неделю, так и с целью остановки кровотечения (симптоматическая гемостатическая терапия — «лечение по требованию» — от 20 до 50 МЕ/кг и далее в зависимости от клинической ситуации). Эти две модели лечения могут чередоваться между собой в течение всей жизни пациента и являются «краеугольным камнем» протокола.
Лечение заболевания может осуществляться не только с целью профилактики кровоизлияний. Протокол ведения больных c гемофилией предусматривает модель лечения состоявшихся кровотечений или кровоизлияний. В этом случае пиковый уровень фактора VIII или IX через 30 минут после инъекции должен составлять 40—100% в зависимости от клинической ситуации.
Инъекции препарата повторяются каждые 12 часов при гемофилии А и каждые 24 часа при гемофилии В в дозе ? от первоначальной до исчезновения симптомов кровоизлияния. Как показывает наш опыт, такая схема лечения приемлема для пациентов с единичными геморрагическими осложнениями. Как и при многих других заболеваниях, при гемофилии легче профилактировать кровоизлияния, а не лечить их осложнения.
Документ общерекомендательного характера
Тем не менее лечащим врачам-гематологам надлежит помнить, что данный документ носит общерекомендательный характер, а подход к лечению каждого пациента должен быть индивидуальным, учитывая порой психоэмоциональный профиль пациента и даже членов его семьи.
Наследственные коагулопатии неизлечимы, но возможности современной препаратной терапии позволяют обеспечить больным продолжительность и качество жизни, сравнимые с таковой в общей популяции.
Внутривенное введение препарата осуществляется самим пациентом или его род-ственниками после прохождения специализированной программы обучения, а наблюдение за пациентом осуществляет центр гемофилии или врач-гематолог.
Такое лечение получило название «домашнее лечение» и используется во всем мире. В нашей стране все больные гемофилией находятся в настоящий момент на «домашнем лечении».
Установлено также, что фенотип заболевания может отличаться от его генотипа. Не все больные с одинаковым уровнем фактора VIII или IX требуют одинакового лечения. 10—15% больных с тяжелой формой гемофилии А вообще не нуждаются в профилактическом лечении, т.е. в постоянном введении препаратов, однако все без исключения больные гемофилией нуждаются в пожизненном обеспечении факторами свертывания крови VIII или IX. Это касается и других наслед-ственных форм коагулопатий — болезни Виллебранда и гипопроконвертинемии (дефицит фактора VII).
Современные антигемофильные препараты факторов свертывания крови VIII и IX, с точки зрения этиотропной терапии, направленной на достижение клинического результата, не обладают между собой значительными различиями. Все они компенсируют уровень недостающего фактора свертывания крови в равной степени. Их активность выражается в стандартных международных единицах (МЕ). Условно они могут классифицироваться на факторы свертывания крови VIII содержащие фактор Виллебранда и не содержащие. Количество и качество фактора Виллебранда может значительно отличаться в них. Существуют препараты, в которых содержание фактора Виллебранда повышено. Есть изолированный фактор Виллебранда, не содержащий фактор VIII.
Рекомбинантные факторы свертывания крови
К препаратам плазмы фактора VIII, не содержащим фактора Виллебранда, количество которого у больных гемофилией не изменено, относятся препараты, прошедшие аффинную хроматографию, а также генно-инженерные (рекомбинантные) факторы свертывания крови VIII (МНН: октоког альфа). Группа препаратов октоког альфа представлена препаратами трех поколений (классификация условна): первое, содержащее добавленный альбумин, необходимый для стабилизации молекулы фактора VIII, второе — содержащее следы человеческого альбумина, и третье — свободное от присутствия альбумина.
Основой эффективной терапии наследственных коагулопатий является: ранняя диагностика, выбор правильной модели лечения и полноценное, беспрерывное обеспечение больных факторами свертывания крови VIII или IX. Если количество препарата недостаточно для лечения, заболевание начинает неуклонно прогрессировать и может свести на нет все ранее предпринятые усилия.
Существует рекомбинантный фактор VIII с модифицированной молекулой, т.е. удаленным гликопротеидным фрагментом (мороктоког альфа). Опыт его использования не так объемен, как у предыдущих препаратов. Для больных с гемофилией В могут быть использованы препараты фактора свертывания крови IX — рекомбинантные (наноког альфа) и полученные из донорской плазмы, а также препараты протромбинового комплекса (PPSB).
У больных с болезнью Виллебранда более целесообразно применять препараты, содержащие физиологическое или больше физиологического соотношения фактора Виллебранда к фактору VIII. Эти препараты были разработаны специально для лечения больных с болезнью Виллебранда, и содержание последнего указывается на этикетке флакона.
Почти абсолютная вирусная безопасность
Накопленный опыт применения рекомбинантных и плазменных препаратов свидетельствует о почти абсолютной вирусной безопасности в отношении вирусов ВИЧ-1, ВИЧ-2, гепатитов В и С. За последние 20 лет не было достоверно зарегистрировано ни одного случая заражения реципиента, однако дискуссии, основанные на «потенциальной угрозе», продолжаются.
Осложнения при гемофилии, как след-ствие лечения, также неизбежны. Наиболее опасное, но редко встречающееся в России (около 3—5% больных гемофилией А) — появление резистентности к терапии, обусловленное образованием иммуноглобулинов, чаще G класса, т.н. аутоантител, которые избирательно блокируют прокоагулянтную активность молекулы фактора VIII. При гемофилии В образование антител отмечается крайне редко (менее 1—2%).
Описаны случаи образования антител и к фактору Виллебранда. К фактору VII антитела практически не образуются. Для практикующих гематологов важно знать, что антигенная стимуляция (введение препарата) может стимулировать иммунный ответ и вызвать полную толерантность к заместительной терапии. Заподозрить ингибитор стоит в случае неэффективности эффективной ранее терапии. Этиология и патогенез этого явления остаются неясными.
Диагностика в регионах недоступна
Терапия и ее мониторинг у больных с гемофилией, осложненной ингибитором, представляет значительные трудности даже для опытных специалистов и лабораторных работников. Принципиально существуют два вида терапии: активация системы гемостаза через шунтирующие (обходные) пути. Для этого используется рекомбинантный активированный фактор VII или протромбиновый комплекс (активированный) — факторы свертывания крови II, VII, IX, X в комбинации. Второй вариант лечения — индукция иммунной толерантности, как правило, «Боннский протокол». В госпитальной практике может использоваться плазмаферез или иммуноадсорбция протеином А, однако эта терапия ведет к временному снижению титра ингибитора и в настоящее время применяется редко. Лечение ингибиторных больных гемофилией представляет серьезную медико-социальную проблему.
При болезни Виллебранда, характеризующейся снижением активности фактора Виллебранда, ристоцетин-индуцированной агрегации тромбоцитов, часто со снижением уровня фактора VIII и антигена фактора VIII, важно определить следующее: имеет ли место недостаток выработки фактора Виллебранда (количественный дефицит — тип I) или синтезируемая структура молекулы фактора неполноценна (качественный дефицит — тип II).
Диагностика заболевания достаточно сложна и часто недоступна в регионах, т.к. требует проведения сложных коагулологических исследований. Важными симптомами заболевания являются длительные носовые кровотечения, у женщин затяжные mensis, которые могут приводить к железодефицитной анемии, длительные кровотечения после удаления зуба или незначительных хирургических вмешательств и проявление таких же симптомов у близких родственников (отец, мать, брат, сестра). Пациенты с подобными симптомами должны быть обследованы в специализированных центрах.
Крайне редкое заболевание
Крайне редко встречающееся заболевание — наследственный дефицит фактора VII. При выраженном дефиците этого фактора отмечается тяжелый геморрагический синдром, сопровождающийся гематомами, гемартрозами, а у женщин — опасными для жизни меноррагиями.
Для лечения больных с гипопроконвертинемией может применяться фактор свертывания крови VII (плазменный) в дозе 30—40 МЕ/кг каждые 8—10 часов. При его отсутствии — эптаког альфа активированный в дозе 20—40 мкг/кг каждые 2—4 часа до полной остановки кровотечения.
Основой эффективной терапии наслед-ственных коагулопатий является: ранняя диагностика, выбор правильной модели лечения и полноценное, беспрерывное обеспечение больных факторами свертывания крови VIII или IX. Если количество препарата недостаточно для лечения, заболевание начинает неуклонно прогрессировать и может свести на нет все ранее предпринятые усилия.
Наблюдение за больными с наследственными коагулопатиями осуществляется в течение всей их жизни в специализированных центрах, часто совместно с врачами других специальностей, если возникают сопутствующие заболевания. Организация подобных центров в России — это организация «технологии лечения» больных, страдающих нарушениями свертывающей системы крови.
Причины плохой свертываемости крови
Исходя из вышеизложенного, можно назвать основные причины нарушений свертываемости:
- Дефекты сосудистой стенки наследственные (связаны с аномалией коллагена) и приобретенные (иммунные или воспалительные поражения сосудов).
- Патология тромбоцитов. Тромбоцитарная дисфункция вызывает иногда значительное кровотечение. Она включает тромбоцитопении (количественные изменения тромбоцитов) и тромбоцитопатии (изменения качества тромбоцитов). Приобретенные тромбоцитопатии вызываются приемом нестероидных противовоспалительных средств, дипиридамола, антибиотиков, уремией, патологией клапанов сердца, использованием экстракорпорального кровообращения.
- Причиной нарушения свертываемости крови может быть недостаток факторов (их тринадцать) свертывающей системы крови или пониженный синтез одного или нескольких факторов. Протромбин (фактор ІІ) — главный компонент свертывания крови. Является предшественником тромбина и участвует в формировании сгустка (тромба). Фибриноген (фактор I) вырабатывается в печени. В каскаде коагуляции он превращается в фибрин, участвующий в образовании сгустка. О дефиците этого фактора указывалось выше.
- Дефицит фактора XI связан с гемофилией С, фактора VIII с гемофилией A, а фактора IX с гемофилией B. Среди наследственных коагулопатий наиболее часто (в 95 % случаев) встречается дефицит факторов VIII и IX. Дефицит VII, X, V, XI факторов составляет только 1,5 %. Приобретенный дефицит факторов протромбинового комплекса (II, VII, X, V) встречается при заболеваниях печени, желтухе, дисбактериозе, а также при передозировке антагонистами витамина К.
- Причиной кровотечений также может быть повышенный фибринолиз, то есть избыточная фибринолитическая активность. Это может быть наследственный дефицит aльфа2-антиплазмина или повышенное образование активаторов плазминогена и нарушение выведения этих активаторов при заболеваниях печени.
Симптомы
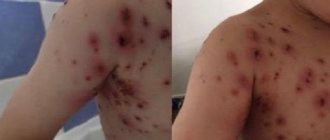
Какой бы ни была причина коагулопатии основным симптомом является кровотечение разной степени выраженности — от небольших синяков до сильных кровотечений во время травм (в том числе и незначительных). Со стороны кожи у больных появляются мелкие петехии, гематомы, синяки в местах инъекций, носовые и десневые, обильные маточные кровотечения у женщин, а также часто желудочно-кишечные кровотечения.
Тромбоцитопатии как врожденные, так и или приобретенные не сопровождаются выраженными геморрагиями. Кровотечение у таких пациентов может развиваться только при операциях, травмах и удалении зубов. Наиболее частые проявления — синяки и периодические носовые и десневые кровотечения.
Тромбоцитопеническая пурпура очень распространённое заболевание, особенно среди женщин 20-30 лет. У больных появляются петехии, кровянистые пузырьки, которые возвышаются над кожей, кровоточивость десен и обильные маточные кровотечения. Заболевание начинается или постепенно или остро с геморрагическим синдромом. По проявлениям бывает два варианта пурпуры: «влажный», когда кровоизлияния сочетаются с кровотечениями и «сухой», если у больного есть только кожные кровоизлияния. Геморрагический синдром на коже отмечается у 100% пациентов.
Количество кровоизлияний может быть единичными и множественными. Для кожного геморрагического синдрома характерно:
- Разные геморрагические высыпания — петехии и крупные кровоизлияния.
- Несоответствие геморрагий степени травмы.
- Спонтанное появление ночью.
- Разный цвет кожных геморрагий в зависимости от давности.
- Безболезненность.
- Асимметрия элементов.
- Кровоизлияния в мягкое небо и миндалины, склеру, глазное дно. Кровоизлияние в склеру иногда предшествует тяжёлому кровоизлиянию в головной мозг, которое возникает быстро и прогрессирует.
- Проявляется головокружением, головной болью, судорогами.
При болезни Виллебранда отмечается склонность к внутрикожным кровоизлияниям, излияниям крови в слизистые и сильные кровотечения после травмы.
Клинические проявления и диагностика дефицита витамина К
В тех случаях, когда на указанные выше этиологические факторы своевременно не обратили внимания, дефицит витамина К нарастает, что в конечном итоге приводит к развитию у ребенка геморрагического синдрома. Как правило, клиническая манифестация ВКДК отмечается после 3–4-й нед. жизни ребенка, наиболее часто это происходит в 1,5–2-месячном возрасте. При этом очень важно помнить о том, что максимальная эффективность лечебных мероприятий при ВКДК достигается в тех случаях, когда терапия начинается при минимальных геморрагических проявлениях [11, 17]. В связи с этим при объективном осмотре необходимо обращать внимание даже на самые незначительные геморрагические симптомы, что станет основанием для уточнения их причин. В этом возрасте геморрагический синдром характеризуется низкой специфичностью, а максимальная эффективность лечения достигается только при этиопатогетическом подходе, поэтому поиск причины становится очень важным. Учитывая, что геморрагический синдром у детей грудного возраста может развиваться катастрофически быстро, необходимо, не теряя времени, собрать анамнез и срочно (по cito!
) выполнить клинический анализ крови с исследованием тромбоцитов и тромбоцитарных индексов, коагулограмму, биохимический анализ крови, определить группу крови и резус-фактор, а также провести нейросонографию из-за риска внутричерепного кровоизлияния (рис. 1).
При сборе семейного анамнеза особое внимание должно быть уделено наличию у ближайших родственников ребенка заболеваний, сопровождающихся геморрагическим синдромом (тромбоцитопении, тромбоцитопатии, коагулопатии, тромбофилии). Очень важно также выяснить, назначены ли ребенку медикаменты, которые могут быть причиной геморрагического синдрома. Кроме этого, в тех случаях, когда ребенок находится на грудном вскармливании, необходимо уточнить, какие лекарственные препараты используются матерью. Обязательно также учитываются такие анамнестические факторы риска, как недоношенность, длительная антибиотикотерапия, парентеральное питание без дотации витаминов. Отдельно необходимо уточнить наличие у ребенка наследственных нарушений метаболизма, врожденных инфекций, пороков развития печени, желчного пузыря, кишечника, а также тяжелых приобретенных заболеваний гепатобилиарной и интестинальной систем (рис. 1).
При осмотре ребенка с геморрагическим синдромом, независимо от характера и степени выраженности клинических проявлений, необходимо обязательно оценить уровень сознания, цвет кожи и видимых слизистых, окраску кала и мочи, состояние периферических лимфоузлов, печени, селезенки. При этом наличие иктеричности слизистых и кожи при одновременном осветлении каловых масс и потемнении мочи позволяет предположить поражение гепатобилиарного тракта с развитием холестаза. В этих случаях в первую очередь необходимо думать о том, что геморрагический синдром, вероятно, обусловлен приобретенными нарушениями вторичного гемостаза, т. к. синтез плазменных факторов системы коагуляции осуществляется в печени.
Отдельного анализа требует детальная характеристика клинических проявлений геморрагического синдрома. Так, в случае повышенной кровоточивости и кровотечения необходимо не только указать локализацию (слизистые, пупочная ранка, желудочно-кишечный тракт, место инъекции и т. д.), но и отметить время, когда впервые появилась указанная симптоматика, а также уточнить, было ли развитие спонтанным или явилось ответом на провоцирующие факторы. При геморрагических проявлениях на коже и/или слизистых следует указать локализацию, распространенность, представить морфологическое описание элементов, выраженность и т. д. Очень важно отметить, когда впервые появились геморрагические проявления, чем, по мнению родителей, они могли быть спровоцированы, указать их продолжительность. Так, нередко при дефиците витамина К в организме первым проявлением ВКДК является длительная кровоточивость из места забора крови для клинического анализа или из места, куда была введена вакцина. С учетом того, что забор крови для гемограммы, как правило, предшествует вакцинации, выявленная при этом кровоточивость должна являться абсолютным показанием для исследования количества тромбоцитов, их индексов и коагулограммы. Вакцинацию при этом проводить нельзя вплоть до получения результатов, исключающих нарушения гемостаза. Особо следует отметить, что наши данные, которые согласуются с результатами исследований других авторов, свидетельствуют о том, что недооценка ранних проявлений геморрагического синдрома и отсутствие адекватной терапии на этом этапе приводят к значительному увеличению частоты тяжелых осложнений из-за развития в дальнейшем внутричерепных кровоизлияний [11, 17–21].
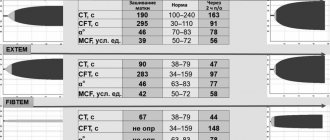
В случае выявления тромбоцитопении в клиническом анализе крови при отсутствии отклонений в коагулограмме необходимо проводить дифференциальную диагностику с целым рядом патологических состояний, при которых отмечается снижение количества тромбоцитов (рис. 2). Если же выявляются изменения в коагулограмме, а все лабораторные показатели первичного гемостаза в клиническом анализе крови (количество тромбоцитов, тромбоцитарные индексы, длительность кровотечения) остаются в пределах нормы, то можно сделать вывод о том, что имеет место коагулопатия (рис. 3). При этом очень важно дифференцировать, на каком этапе каскадной системы гемостаза имеется сбой. Для этого целесообразно обратить внимание на то, что при гемофилиях отмечается удлинение активированного частичного времени (АЧТВ) при нормальных показателях «внешнего пути» (протромбиновый индекс (ПТИ), протромбиновое время (ПВ), международное нормализованное отношение (МНО)) и конечного этапа свертывания (тромбиновое время (ТВ), фибриноген). Для селективного дефицита VII фактора свертывания характерны изменения показателей «внешнего пути» (снижение ПТИ, удлинение ПВ и увеличение МНО) при нормальных значениях показателей «внутреннего пути» (АЧТВ) и конечного этапа свертывания (ТВ, фибриноген). Для а- или гипофибриногенемии типичными изменениями в коагулограмме являются удлинение АЧТВ и ТВ, уменьшение ПТИ, повышение ПВ и МНО, снижение уровня фибриногена [22].
В тех же случаях, когда при отсутствии изменений в тромбоцитарном звене выявляются нарушения во «внутреннем пути» (удлинение АЧТВ или полное отсутствие коагуляции) и «внешнем пути» (снижение ПТИ, повышение ПВ, увеличение МНО или полное отсутствие коагуляции), в то время как показатели конечного этапа свертывания (ТВ, фибриноген) остаются в пределах нормы, следует предположить ВКДК (рис. 4). Указанные особенности коагулограммы при ВКДК обусловлены тем, что витамин-К-зависимые факторы свертывания крови представлены на разных этапах «внутреннего пути» и «внешнего пути» коагуляции, но не принимают участия в конечном этапе. Так, IX фактор свертывания является обязательным компонентом «внутреннего пути», поэтому его дефицит приведет к удлинению АЧТВ или полному отсутствию коагуляции при глубоком дефиците. В свою очередь при недостаточности VII фактора свертывания, являющегося ключевым инициатором активации «внешнего пути», отмечается снижение ПТИ, повышение ПВ, увеличение МНО или полное отсутствие коагуляции при постановке этих проб, если имеет место глубокий дефицит VII фактора. II и X факторы свертывания принимают участие в обоих путях свертывания, их дефицит также будет сопровождаться изменениями показателей, характеризующих как «внутренний путь» (удлинение АЧТВ или полное отсутствие коагуляции при глубоком дефиците), так и «внешний путь» (снижение ПТИ, повышение ПВ, увеличение МНО или полное отсутствие коагуляции). В то же время во всех этих случаях показатели конечного этапа свертывания (ТВ, фибриноген) остаются в пределах нормы, т. к. витамин-К-зависимые факторы свертывания не принимают участия в коагуляции на этом уровне (рис. 4).
Анализы и диагностика
Коагулологический скрининг включает:
- протромбиновый индекс;
- активированное частичное тромбопластиновое время;
- количество фибриногена;
- количество тромбоцитов;
- время кровотечения.
При изолированном удлинении активированного частичного тромбопластинового времени переходят на второй этап обследований:
- делают тест коррекции;
- активность факторов VIII, IX, XI, ХII.
При снижении активности фактора VIII переходят к третьему этапу обследований:
- волчаночный антикоагулянт;
- специфический ингибитор фактора VIII.
У детей
Плохая свертываемость крови у ребенка часто связана с иммунной тромбоцитопенической пурпурой. Острая пурпура развивается в возрасте 2-9 лет. Это иммуно обусловленное заболевание, характеризующееся постоянным (или периодическим) снижением тромбоцитов меньше 100 тысяч. Нарушение у детей возникает через 1-3 недели после вирусной инфекции. Запуститься такая патологическая реактивность может не только под влиянием вирусной инфекции, но и после приёма препаратов, проведения вакцинации, воздействия температур (как низких, так и высоких), оперативных вмешательств или эмоционального перенапряжения. На фоне нормального самочувствия у ребенка появляются петехиальная сыпь (на слизистых и коже), синяки повторяющиеся носовые кровотечения и кровоточивость десен. В тяжелых случаях могут быть кровоизлияния в мозг и желудочные кровотечения. Так как антигены вируса постепенно уходят из крови, у большинства заболевание через 2 месяца самостоятельно проходит.
У ребенка первых месяцев жизни и новорожденных встречается витамин-К-дефицитная коагулопатия. При недостаточности Vit-К снижается активность некоторых факторов: протромбин, проконвертин, фактор Кристмаса и фактор Прауэра. В результате развивается гипокоагуляция, сопровождающаяся геморрагическим синдромом.
Причины дефицита Vit-К у новорожденного:
- прием во время беременности антикоагулянтов;
- антибиотиков;
- противосудорожных препаратов;
- тяжелые поражения печени у беременной и кишечника;
- наличие фетоплацентарной недостаточности;
- гестоз и преэклампсия.
Проявления этой коагулопатии у новорожденных малоспецифичны — кожный синдром, повышенная кровоточивость при заборе крови и кровотечение из пупочной ранки. При недостатке Vit-К длительность кровоточивости и уровень тромбоцитов в пределах нормы. Многие авторы рекомендуют профилактически всем детям сразу после рождения вводить Vit K — 2-3 введения первые 1,5 месяца, а в некоторых случаях еженедельное введение продолжается до 3-х месяцев.
В более позднем возрасте Vit-К-дефицитная коагулопатия обусловлена вскармливанием только грудным молоком. При этом в 78% у детей появляются массивные внутричерепные кровоизлияния. Значительно реже причиной сниженной свертываемости у детей является ДВС-синдром при тяжелом сепсисе, врожденные изменения обмена веществ и наследственные коагулопатии.
Этиология витамин-К-дефицитной коагулопатии у детей в постнеонатальном периоде
ВКДК является одной из ведущих причин геморрагического синдрома у детей первых месяцев жизни [1–11]. Особо следует отметить, что ВКДК, развивающаяся у детей в постнеонатальном периоде, по-прежнему обозначается термином «поздняя геморрагическая болезнь новорожденного». При этом отмечено, что указанный подход не только приводит к терминологической путанице, но и определяет диагностические ошибки. Так, ранее нами при анонимном анкетировании 348 врачей-педиатров было показано, что позднюю геморрагическую болезнь новорожденного подавляющее большинство респондентов (2/3) связывали исключительно с неонатальным периодом и демонстрировали низкий уровень информированности о клинических проявлениях, методах диагностики и лечения ВКДК у детей грудного возраста [7]. Причиной указанных ошибок явилось ложное представление респондентов о том, что дефицит витамина К имеет четкие возрастные интервалы и ограничен периодом новорожденности. Более того, практически во всех случаях анкетируемые врачи-педиатры считали, что поздняя геморрагическая болезнь новорожденного обусловлена отсутствием профилактического введения витамина К в роддоме и не связана с такими состояниями, как вскармливание исключительно материнским молоком, билиарная атрезия, тяжелые поражения гепатобилиарной системы или кишечника, наследственные заболевания, сопровождающиеся холестазом [7].
Учитывая это, мы предложили исключить из обращения такое понятие, как «поздняя геморрагическая болезнь новорожденного», заменив его термином «ВКДК» [7]. Указанный подход позволит дифференцировать имеющие одинаковый патогенез (дефицит витамина К и обусловленная этим коагулопатия), но различающиеся по этиологии и срокам манифестации геморрагические синдромы. Так, при геморрагической болезни плода и новорожденного (код Р53 по МКБ-10) дефицит витамина К развивается в перинатальный период и обусловлен недостаточным его трансплацентарным поступлением. При этом варианте ВКДК клиническая манифестация отмечается в первые 1–7 дней жизни. Следует особо подчеркнуть, что введение витамина К ребенку сразу после рождения с высокой эффективностью купирует перинатальный дефицит витамина К и в подавляющем большинстве случаев предупреждает развитие геморрагического синдрома [1–5, 10–12]. В отличие от этого развитие ВКДК в постнеонатальный период, которую традиционно называют поздней геморрагической болезнью новорожденного, обусловлено целым рядом причин (вскармливание исключительно грудным молоком, пороки развития или тяжелые приобретенные поражения гепатобилиарной системы и/или кишечника, наследственные заболевания, сопровождающиеся холестазом, длительное применение антибиотиков и пролонгированное парентеральное питание с недостаточной дотацией витамина К) и не предупреждается однократным введением витамина К сразу после рождения [1–5, 7–11, 13–17]. Учитывая это, следует признать ошибочным мнение, что назначение витамина К в раннем неонатальном периоде профилактирует развитие ВКДК в последующие месяцы жизни ребенка. В связи с этим при обсуждении данного вопроса очень важно подчеркивать, что дефицит витамина К в постнеонатальном периоде обусловлен совсем другими причинами. При этом раннее их выявление позволяет своевременно провести заместительную терапию, что корригирует дефицит витамина К и предупреждает развитие ВКДК.
Плохая свертываемость крови при беременности
Нормально протекающая беременность всегда сопровождается важными биохимическими изменениями, в том числе и в системе гемостаза. Однако встречаются патологические состояния, приводящие к нарушению свертываемости крови. Коагулопатия беременных, что это? Это патологическое состояние, протекающее с нарушением свёртывания и повышенным риском кровотечений.
Патологическая кровоточивость у беременных может быть обусловлена:
- Врожденными нарушениями в системе свертывания.
- Болезнью Верльгофа.
- Болезнью Мошковица.
- Дефицитом антитромбина, который часто встречается при беременности.
- Преэклампсией.
- Эклампсией.
- ДВС-синдромом, связанном с преэклампсией. На первом этапе возникает внутрисосудистое свертывание крови, а потом происходит истощение свертывающей системы.
- HELLP-синдромом, который тоже связан с преэклампсией (гемолиз, повышенный уровень печеночных ферментов и тромбоцитопению). Синдром диссеминированного внутрисосудистого свертывания крови, сочетание тромбоза с кровотечениями.
- HELLP-синдром сочетает триаду проявлений: гемолиз, снижение уровня тромбоцитов и повышение печеночных ферментов. При беременности с тяжелой преэклампсией частота этого синдрома достигает 20%.
- Он развивается при доношенной беременности, преждевременных родах и даже после родов. HELLP-синдром считается подтипом преэклампсии. Диагностические критерии этого синдрома: тромбоциты менее 100×10 в 9/л, трансаминазы выше нормы в 2-3 раза, гемолиз эритроцитов, билирубина более 20,5 мкмоль/л.
У женщин появляется тошнота, отёки и боли в правом подреберье. Беременным с HELLP-синдромом назначают инъекции сульфата магния до родов и после родов в течение двух суток. Трансфузия тромбоцитов показана при тромбоцитах меньше 20×10 в 9/л, если предстоят естественные роды и при тромбоцитах меньше 50×10 в 9/л, если предстоит кесарево сечение. Тромбоконцентрат вводят перед родоразрешением. Кортикостероиды увеличивают уровень тромбоцитов, поэтому их назначение обоснованно. В послеродовом периоде применяется плазмообмен.
При ДВС-синдроме недлительная фаза гиперкоагуляции сменяется гипокоагуляцией. Такие изменения происходят если женщина теряет 15-20% объема крови. При кровотечении производят инфузию тромбоконцентрата.
При кровотечении и, если повышены протромбиновое время и активированное тромбопластиновое время проводится инфузия свежезамороженной плазмы. Если ввести плазму нет возможности, вводят концентраты факторов свертывания. Тяжелая гипофибриногенемия, которая не корректируется переливанием плазмы, тогда переливают криопреципитат. При гиперфибринолизе и кровотечении вводится транексамовая кислота. Гемолитико-уремический синдром у женщин сопровождается тромбоцитопенией, поражением почек и микроангиопатией. Это состояние чаще развивается после родов. Плазмообмен при данной патологии не очень эффективен — женщине требуется гемодиализ.
Лечение витамин-К-дефицитных состояний
Своевременное установление причины геморрагического синдрома позволяет назначить адекватную этиопатогенетическую терапию на ранних этапах заболевания, что помогает не только купировать патологический процесс, но и предупредить развитие осложнений. В тех случаях, когда речь идет о ВКДК у детей первых месяцев жизни, следует помнить о том, что в 50–75% случаев ВКДК приводит к внутричерепным кровоизлияниям, сопровождающимся высокой частотой неблагоприятных исходов, а у выживших детей — серьезными осложнениями [1–5, 7–11, 13–21]. При этом большинство авторов подчеркивают, что внутричерепные кровоизлияния на фоне дефицита витамина К развиваются спустя некоторое время после появления кожных и/или слизистых геморрагий, а в ряде случаев — на фоне продолжающегося кровотечения из места забора крови или инъекции, которые не были своевременно замечены или оставались недооцененными. В связи с этим еще раз целесообразно подчеркнуть необходимость экстренного поиска причин даже при незначительной выраженности геморрагических проявлений. В тех случаях, когда на основании анализа клинико-анамнестических данных и результатов лабораторного обследования верифицирована ВКДК, необходимо незамедлительно начинать заместительную терапию. Препаратами выбора при этом являются так называемые протромбиновые комплексы — лекарственные средства, в составе которых содержатся все витамин-К-зависимые факторы свертывания (II, VII, IX, X), а также белки С и S. После купирования геморрагического синдрома оправдано плановое назначение витамина К. В тех случаях, когда нет возможности использовать препараты протромбинового комплекса, вводят одногрупную свежезамороженную плазму (10 мл/кг массы тела) и витамин К. При этом следует помнить, что если эффект от заместительной терапии препаратами протромбинового комплекса или свежезамороженной плазмы наступает уже в период их введения, то единственный зарегистрированный в нашей стране синтетический аналог витамина К (менадиона натрия бисульфит) проявит свой гемостатический эффект только через 18–24 ч. В связи с этим при развитии геморрагического синдрома, обусловленного ВКДК, нельзя ограничиваться только введением менадиона натрия бисульфита — требуется одновременное применение препаратов, содержащих витамин-К-зависимые факторы (или протромбиновый комплекс, или свежезамороженная плазма).
Профилактика
Повлиять на врожденные патологии свертывающей системы невозможно, но лица, имеющие такую патологию, могут более тщательно подходить к своему здоровью, выбору профессии и физической активности. Вторичная профилактика включает следующее:
- Избегать физической нагрузки, связанной с возможностью получить ушибы (спортивные состязания, занятия футболом, борьбой, фигурным катанием и прочее).
- Гигиена полости рта, которая помогает снизить потребность в стоматологических процедурах и операциях.
- Нормализовать вес, который дает нагрузку на суставы и повышает риск кровоизлияний в полость сустава.
- Избегать применения медикаментов, которые оказывают влияние на свертываемость. Среди таких препаратов можно назвать ацетилсалициловую кислоту, Клопидогрель, Кофеин, Ибупрофен, Напроксен, нитрофураны, барбитураты, карбенициллин.
- Женщины с врожденными дефицитами факторов должны проконсультироваться и обследоваться у генетика и выяснить риски рождения ребенка с врожденной патологией свертывания крови.
Точно также отсутствует первичная профилактика тромбоцитопенической пурпуры, а вторичная сводится к предупреждению обострений. Больные не должны пребывать на солнце, им противопоказана работа в условиях повышенной температуры (горячий цех, пищевой цех у плиты). Детей освобождают от физкультуры. После каждого случая ОРВИ в обязательном порядке нужно исследовать кровь.
Последствия и осложнения
При коагулопатиях с разной вероятностью и разной выраженностью могут отмечаться следующие осложнения:
- Анемия.
- Гематурия (кровь в моче).
- Обильные и продолжительные месячные.
- Кровоизлияние в мозг.
- Желудочно-кишечные кровотечения.
- Кровоизлияние в структуры глаза, плевриты, сдавление гортани и трахеи гематомами.
- Кровоизлияние в суставы и как следствие этого развитие артритов, артрозов, остеопороза и постгеморрагических бурситов.
- Внутрипозвоночные кровоизлияния.
- В редких случаях смерть при массивных кровотечениях.
Прогноз
Острые формы идиопатической пурпуры проходят течение нескольких месяцев и часто бывает так, что заболевание не рецидивирует. При хронической форме удается вызвать ремиссию, но болезнь часто рецидивирует.
При использовании современных медикаментов и выполнении всех рекомендаций по изменению образа жизни и трудоустройства тромбоцитопеническая пурпура имеет благоприятный прогноз.
Гемофилия без лечения приводит к инвалидности и порой к преждевременной смерти. Отсутствие лечения часто приводит к патологии суставов (гемофилическая артропатия), что требует потом использования костылей и колясок или специального ортопедического лечения. На фоне лечения продолжительность жизни практически не отличается от здоровых лиц.
Список источников
- Фаткуллин И.Ф., Зубаиров Д.М. Наследственные и приобретенные дефекты системы гемостаза в акушерско-гинекологической практике. М., 2002. С. 64.
- Галстян Г.М., Суханова Г. А. Введение в гемостаз, современные препараты крови и их влияние на коагуляцию// Медицинский Совет.2013. с 11-13.
- Баркаган З.С. Диагностика и контролируемая терапия нарушений гемостаза / З.С. Баркаган, А.П. Момот. – М.: Ньюдиамед, 2001. – 296 с.
- Баринов С. В., Долгих В. Т., Медянникова И.В. Гемокоагуляционные нарушения у беременных с гестозом. Журнал акушерства и женских болезней. – 2013; 62 (6): 5–12.
- Дегтярев Д. Н., Карпова А. Л., Мебелова И.И. и др. Проект клинических рекомендаций по диагностике и лечению геморрагической болезни новорожденных // Неонатология. 2015. № 2. C. 75–86.
Эмболия околоплодными водами
Этот комплексное расстройство характеризуется внезапным развитием гипотензии, гипоксии и коагулопатии потребления и имеет вариабельный клиническую манифестацию. При родоразрешении или сразу после родов у матери развивается одышка, судороги, остановка дыхания и сердечной деятельности, усложняется диссеминированной внутрисосудистой коагуляцией, массивным кровотечением и завершается смертью.
Патогенез. Амниотическая жидкость попадает в микроциркуляцию вследствие нарушения физиологического барьера, который в норме существует между материнской и плодовой сосудистой системой. Воротами для попадания элементов тканей плода в материнскую сосудистую систему могут быть травма, амниоцентез, но чаще всего это разрывы шейки матки и нижнего маточного сегмента во время родов. Кесарево сечение также предоставляет возможность смешивания материнской крови и плодовых тканей.
Патофизиологический каскад запускается многочисленными хемокинами и цитокинами. После короткой начальной фазы легочной и общей гипертензии происходит уменьшение периферической сосудистой резистентности и ударного рабочего объема левого желудочка. Транзиторная, но глубокая дезатурация кислорода в начальную фазу приводит к неврологическим осложнениям у тех больных, что выживают. У тех, кто выживает после начального сердечно-сосудистого коллапса, развивается вторичная фаза поражения легких и коагулопатии потребления.
Гипертонус матки не является причиной эмболии, как часто считают. Маточный кровоток полностью прекращается при росте внутриматочного давления до 35-40 мм рт. ст. Итак, гипертонические сокращения матки является наименее вероятным временем для фетоматеринского обмена. Согласно данным современных исследований, применение окситоцина не имеет связи с увеличением случаев эмболии околоплодными водами.
Диагностика эмболии околоплодными водами раньше базировалась на нахождении клеток плоского эпителия плода в центральных легочных сосудах матери, считалось патогномоническим признаком эмболии. Современные исследования показывают, что плоские клетки плода, трофобласт и другие продукты плодового происхождения могут находиться в материнской сосудистой системе без связи с эмболией околоплодными водами. Итак, наличие плодовых тканей в легочных сосудах не является ни чувствительным, ни специфичным диагностическим тестом. Диагноз эмболии околоплодными водами определяется методом исключения других причин смерти.
Лечение эмболии околоплодными водами состоит в стабилизации жизненных функций (легочно-сердечная реанимация), восстановлении ОЦК и лечении нарушений коагуляции. Но нет достоверных данных, что любое вмешательство улучшает прогноз при эмболии околоплодными водами. Если эмболия околоплодными водами имеется к родоразрешению, рекомендуют ургентное кесарево сечение с целью спасения жизни плода. Но при условии нестабильности гемодинамики матери возможность такого вмешательства усложняется.
Если больной выживает, прогноз для матери и плода осложняется возможностью серьезных постгипоксических неврологических расстройств. Последствия для плода ухудшаются при увеличении интервала от эпизода эмболии до родоразрешения.