Myocardial hypertrophy: causes of occurrence
The mechanism of myocardial hypertrophy is quite simple. Everyone knows that the heart performs a “pumping” function, pumping all the blood in the body, delivering oxygen to every organ. Constant excessive physical and emotional stress leads to an increased need for oxygen in all organs and, consequently, to a large volume of blood. All this leads to the fact that the myocardium cannot cope with its work, loses its elasticity and increases in size. Myocardial hypertrophy usually occurs for the following reasons: - arterial hypertension; — acquired and congenital heart defects; — disturbance of blood supply; - obesity; - constant overload on the heart; - genetic predisposition... It should also be noted that myocardial hypertrophy most often affects children and this disease is a sign of congenital heart disease.
Left ventricular hypertrophy of unknown etiology
S.V. Moiseev. Left ventricular hypertrophy (LVH) is often encountered in cardiologist practice. It can be suspected on the basis of an ECG, but more reliable methods for assessing myocardial mass are echocardiography and especially magnetic resonance imaging (MRI). In echocardiography, the criterion for LVH is considered to be an increase in the mass index of the left ventricular myocardium, respectively, >115 g/m2 in men and >95 g/m2 in women (adjusted for body surface area) or >50 g/m2.7 in men and >47 g/m2.7 in women (adjusted for height) [1], and with MRI - >85 g/m2 in men and >81 g/m2 in women [2]. Depending on the relative wall thickness (RWT) of the left ventricle [(2 × posterior wall thickness)/end diastolic size of the left ventricle], concentric (RWW≥0.43) and eccentric (RWW<0.43) LVH are distinguished. Given the possible heterogeneity of left ventricular thickening, in particular asymmetric increase in the thickness of the interventricular septum or left ventricular apex, an increase in septal or wall thickness of ≥12 mm should be considered when diagnosing LVH.
In the vast majority of patients, the cause of LVH is pressure or volume overload of the heart due to arterial hypertension or heart defects, primarily aortic, but in some patients there are no obvious causes of LVH. The prevalence of unexplained LVH in adults in the general population is 0.02–0.23% [3]. The absence of obvious causes of myocardial hypertrophy usually serves as the basis for a diagnosis of hypertrophic cardiomyopathy (HCM), although in 5-10% of patients LVH of unknown etiology is caused by other genetic and non-genetic diseases, including lysosomal storage diseases (Fabry, Danon, Pompe diseases), ATTR - and AL amyloidosis, neuromuscular diseases (Friedreich’s ataxia), mitochondrial cardiomyopathies, etc. Timely diagnosis of some of them, in particular Fabry and Pompe diseases, AL and ATTR amyloidosis, is of great practical importance, given the possibility of pathogenetic treatment. When making a differential diagnosis, one should take into account the patient's age, the severity of LVH and clinical symptoms, the presence of a family history and various extracardiac manifestations. Some genetic diseases, such as Fabry disease and ATTR amyloidosis, can sometimes only be diagnosed through screening tests. The most common causes of LVH of unknown etiology will be discussed below.
E.V. Privalova. HCM is a hereditary disease that is transmitted in an autosomal dominant manner [4]. According to the recommendations of the European Society of Cardiology 2014, HCM can be diagnosed in the presence of myocardial hypertrophy ≥15 mm in at least one segment of the left ventricle, which cannot be explained by other causes, and in relatives of the patient with an established diagnosis - in the presence of myocardial hypertrophy ≥13 mm [3]. Myocardial hypertrophy in HCM can be either asymmetric (Fig. 1) or symmetric. In 40-60% of patients with HCM, mutations are detected in genes encoding cardiac sarcomere proteins, primarily beta-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3). Less common are mutations in the genes encoding troponins I and T (TNNI3, TNNT2), tropomyosin α1 chain (TPM1), and myosin light chain 3 (MYL3). In general, patients with mutations in sarcomeric protein genes have a higher incidence of family history of HCM and sudden death and more severe LVH and myocardial fibrosis than patients without mutations [5]. When examining a patient, it is important to assess the presence of left ventricular outflow tract obstruction [6]. Its criterion is the pressure gradient in the outflow tract, which is measured by the Doppler method, ≥30 mm Hg. Art. at rest or after provocative tests (Valsalva maneuver, physical activity). An increase in this indicator of ≥50 mm Hg is considered hemodynamically significant. Art. [3].
Rice. 1. Severe asymmetric hypertrophy of the interventricular septum in HCM
V.Yu. Kaplunova. Patient K., 45 years old, was examined at the A.A. Hospital Therapy Clinic. Ostroumova in October 2021. The patient's older brother died suddenly at the age of 54 years. HCM was diagnosed in another brother, who died at age 54, and in the patient's 29-year-old daughter. From the age of 18, the patient had a systolic murmur on the left edge of the sternum in the absence of clinical manifestations and good tolerance to physical activity. At the age of 27 years, shortness of breath, palpitations, interruptions in heart function, dizziness and lightheadedness, and discomfort in the cardiac region with moderate physical activity appeared. At the age of 33 years, asymmetric LVH was discovered (thickness of the interventricular septum - 20 mm, posterior wall - 12 mm) with signs of obstruction of the left ventricular outflow tract and a resting pressure gradient of 45 mm Hg. Art. An obstructive form of HCM was diagnosed. Since the age of 44, he has noticed paroxysms of atrial fibrillation with the subsequent transition of the arrhythmia to a permanent form. Echocardiography revealed an increase in hypertrophy of the interventricular septum to 31 mm and an increase in the pressure gradient to 94 mm Hg. Art. at rest. At the Scientific Center for Cardiovascular Surgery named after. A.N. In Baku, a myectomy was performed using access from the right ventricle, which led to a decrease in the degree of obstruction of the left ventricular outflow tract and diastolic dysfunction. A molecular genetic study revealed a missense mutation in exon 22 of the gene encoding the β myosin heavy chain (MYHT A870C) in the proband and his daughter.
The presented observation illustrates the typical manifestations and course of HCM: asymmetric hypertrophy of the interventricular septum, which was detected at a young age, slowly increased and was not accompanied by clinical symptoms for a long time, in particular congestive heart failure, obstruction of the outflow tract of the left ventricle with a high pressure gradient in its cavity, family history (diagnosis of HCM and/or cases of sudden cardiac death in close relatives), mutation of the gene encoding sarcomeric protein, which was detected in both the proband and his daughter. Treatment of HCM usually begins with β-blockers, which do not have vasodilating activity, which reduce the pressure gradient in the left ventricular cavity and clinical symptoms. If they are ineffective, disopyramide or verapamil can be used. In case of severe hypertrophy of the interventricular septum and a high pressure gradient in the left ventricle, myectomy can be performed, which in more than 90% of cases allows eliminating or significantly reducing outflow tract obstruction, improving exercise tolerance and survival [7].
E.V. Privalova. Differential diagnosis of HCM with other diseases accompanied by LVH can be difficult, for example, in the presence of moderate myocardial hypertrophy, especially symmetrical, not accompanied by obstruction of the left ventricular outflow tract, and in the absence of a family history. Mutations of sarcomeric protein genes are not detected in all patients with HCM, and in some patients with unexplained LVH, molecular genetic testing is not performed for economic reasons. It should be taken into account that asymmetric hypertrophy of the interventricular septum, characteristic of HCM, also occurs in other diseases, including secondary myocardial hypertrophy in arterial hypertension. The cause of LVH can be not only pressure overload of the left ventricle, but also physical training, although in a large study, an increase in the thickness of the left ventricular wall of more than 12 mm was detected in only 1.7% of 947 athletes involved in various sports, and the wall thickness did not exceed 16 mm. A more common echocardiographic sign of “athlete's heart” was dilatation of the left ventricular cavity, which was detected in 38% of cases [8]. Important for the diagnosis of some diseases accompanied by LVH is a thorough analysis of the clinical picture, which makes it possible to identify certain extracardiac manifestations of the disease that are absent in HCM. A myocardial biopsy is not required to confirm the diagnosis of HCM, but histological examination may be reasonable to exclude infiltrative diseases accompanied by thickening of the left ventricular wall.
E.A. Karovaikina. Fabry disease is a rare disease characterized by a disorder of glycophospholipid metabolism due to deficiency or absence of the lysosomal enzyme α-galactosidase A [9]. The cause of deficiency of this enzyme is mutations of the GLA gene located on the X chromosome, therefore typical clinical manifestations of Fabry disease are observed more often and more pronounced in hemizygous men, but they are also often found in heterozygous women. In the classic phenotype of Fabry disease, the first symptoms, in particular neuropathic pain (episodes of burning pain in the hands and feet that occur with fever, exercise, stress and rapid changes in ambient temperature), angiokeratomas (superficial angiomas localized on the anterior abdominal wall, in particular inside or around the navel, in the groin area, on the buttocks, upper limbs, lips; Fig. 2), decreased or absent sweating, gastrointestinal disorders, appear in childhood or adolescence, and at the age of 20-40 years, damage to internal organs develops , including the heart, kidneys (proteinuria and progressive decrease in glomerular filtration rate) and the central nervous system (transient ischemic attacks and stroke). In the atypical “cardiac” variant of the disease, LVH develops at the age of 40-50 years and older in the absence of early symptoms. The results of several large screening studies suggest that pathogenic mutations of the GLA gene, associated with the development of Fabry disease, can be detected in 0.5-1% of patients diagnosed with HCM [10].
Rice. 2. Angiokeratomas in the umbilical area in a patient with Fabry disease
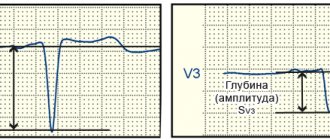
A.S. Moiseev. The following two observations can serve as an illustration of LVH caused by Fabry disease. Patient L., 48 years old, was examined at the clinic named after. EAT. Tareev in October 2021. From the age of 19, severe lymphedema of the lower extremities and decreased sweating. At the age of 47 years, rare pain in the chest appeared, not associated with physical activity, and echocardiography revealed a thickening of the interventricular septum and the wall of the left ventricle up to 14 mm in the absence of dilatation of the heart chambers and disturbances in systolic function. He was seen by a cardiologist with a diagnosis of HCM. After 1 year, screening revealed a decrease in α-galactosidase A activity in dried blood spots, an increase in the level of globotriazylsphingosine (Lyso-GL3) to 117 ng/ml (normal <1.8) and a mutation in the GLA gene (c.145C>G ), which allowed us to establish the diagnosis of Fabry disease. The ECG showed signs of LVH (Fig. 3). Cardiac MRI revealed an increase in the left ventricular myocardial mass index to 123 g/m2 and signs of intramyocardial fibrosis. In addition, there were symptoms of kidney damage - a decrease in the estimated glomerular filtration rate to 62 ml/min/1.73 m2 in the absence of proteinuria.
Rice. 3. ECG signs of myocardial hypertrophy in a patient with Fabry disease
At the same time, we examined the patient’s 67-year-old mother, who, 2 years before hospitalization at the clinic, was also diagnosed with unexplained LVH, accompanied by a persistent form of atrial fibrillation, frequent ventricular extrasystole and heart failure, and therefore took β-blockers, diuretics, digoxin , anticoagulants. Cardiac MRI revealed thickening of the interventricular septum (19 mm) and the posterior wall of the left ventricle (12 mm), an increase in the left ventricular myocardial mass index to 141 g/m2, and areas of intramyocardial fibrosis. MRI of the brain revealed numerous lesions in the white matter, and upon examination by an ophthalmologist, infundibular keratopathy characteristic of Fabry disease, the diagnosis of which was confirmed by the results of a molecular genetic study (mutation c.145C>G), an increase in the level of Lyso-GL3 to 23 ng/ml and decreased activity of α-galactosidase A. The patient and his mother have been receiving enzyme replacement therapy for 1.5 years.
The presented observations demonstrate the importance of screening in the diagnosis of Fabry disease in patients with unexplained LVH, since it was difficult to clinically suspect the correct diagnosis due to the absence of typical early symptoms - neuropathic pain and angiokeratoma. To diagnose Fabry disease in men, it is necessary to determine the activity of α-galactosidase A and/or the level of Lyso-GL3 in dried blood spots. In women, enzyme activity often remains normal or decreases slightly, so determining the level of Lyso-GL3 is considered more informative. To confirm the diagnosis, a molecular genetic study is performed to identify a pathogenic mutation of the GLA gene (in the Russian Federation, all these studies are performed free of charge).
N.R. Nosova. When analyzing the clinical picture, attention was drawn to the late development of LVH and its presence not only in the proband, but also in his mother. As stated above, damage to internal organs in Fabry disease, unlike some other hereditary diseases, is usually observed in men at the age of 30-40 years, and in women at an older age. Inheritance of Fabry disease is linked to the X chromosome, so the patient's mother usually exhibits some manifestation of the disease, although its phenotype may differ. The proband, from the age of 18, had decreased sweating, which occurs in most men with Fabry disease, and lymphedema. According to the Fabry Outcomes Survey registry (n=714), the incidence of lymphedema in this disease was 16% in men and 6% in women [11]. The proband's mother was diagnosed with vortex keratopathy (brown-golden deposits in the cornea in the form of wavy lines emanating from one central point), which is one of the clinical criteria for the diagnosis of Fabry disease. According to our data, the incidence of vortex keratopathy in 69 adult patients with Fabry disease was 65.2%, including 56.4% in men and 76.7% in women [12]. Unlike HCM, myocardial hypertrophy in both the proband and his mother was moderate and was not accompanied by obstruction of the left ventricular outflow tract. Adult patients with Fabry disease usually have damage not only to the heart, but also to the kidneys (albuminuria/proteinuria, decreased glomerular filtration rate) and brain (focal changes in white matter on MRI, transient ischemic attacks/stroke).
E.A. Karovaikina. For Fabry disease, replacement therapy is carried out with recombinant α-galactosidase A drugs (agalsidase alfa at a dose of 0.2 mg/kg or agalsidase beta at a dose of 1 mg/kg), which cause regression of LVH or at least slow down the progression of cardiomyopathy. According to D. Ger main et al., in patients with Fabry disease who began treatment at a younger age (18-30 years), the average mass of the left ventricular myocardium decreased by 3.6 g per year, while without treatment in for men of the same age, it increased by 9.5 g per year (p <0.0001) [13].
V.V. Rameev. The cause of thickening of the heart walls may be not only hypertrophy of cardiomyocytes, but also infiltration of the myocardium with an insoluble fibrillar glycoprotein, amyloid. Currently, about 30 amyloidogenic proteins are known, but more than 95% of cases of cardiac amyloidosis are caused by AL amyloidosis or, less commonly, transthyretin (ATTR) amyloidosis. AL amyloidosis develops with the deposition of monoclonal immunoglobulins in patients with lymphoplasma cell dyscrasias, including multiple myeloma. ATTR amyloid (mutant and wild type) is formed from transthyretin, which is synthesized mainly by the liver and functions as a transport protein for thyroxine and vitamin A. The cause of the development of wild type ATTR amyloidosis (senile) is considered to be an age-related decrease in the activity of hepatocyte enzyme systems, which leads to predominantly secretion of unstable monomeric forms of transthyretin, which easily aggregate in tissues to form amyloid. Hereditary ATTR amyloidosis is based on mutations in the TTR gene, accompanied by the synthesis of transthyretin, which is not capable of forming tetramers and has a very high amyloidogenicity.
A.S. Rameeva. Patient B., 50 years old, was examined for the first time at the clinic named after. EAT. Tareev in January 2015. For a year, she suffered from pain in the heart area and increasing heart failure. Echocardiography revealed thickening of the interventricular septum and posterior wall of the left ventricle up to 14 mm, and restrictive diastolic dysfunction. Coronary angiography revealed moderate stenosis of the right coronary artery (50%). During an examination at the Russian Cardiology Research Center in December 2014, slight proteinuria (0.14 g/l), a decrease in the voltage of the ECG waves (Fig. 4), an increase in the thickness of the interventricular septum and the posterior wall of the left ventricle up to 18 mm, and dilatation of the left atrium with absence of left ventricular dilatation and decreased ejection fraction; cardiac MRI shows diffuse, uneven subendocardial accumulation of contrast agent. Amyloidosis was highly suspected, the diagnosis of which was confirmed by myocardial biopsy. When examining blood using the Freelite method, a sharp increase in the concentration of free lambda type light chains was noted to 1383 mg/l (normally 5.7-26.3 mg/l), indicating the presence of AL amyloidosis. A trephine biopsy diagnosed multiple myeloma (an increase in the number of plasma cells to 20%). The patient was treated with bortezomib, melphalan and dexamethasone, which normalized the concentration of free lambda light chains and achieved compensation for heart failure.
Rice. 4. Low voltage of QRS complexes in the chest leads in amyloidosis of the heart
V.V. Rameev. Thus, the patient was diagnosed with AL amyloidosis with cardiac involvement as part of multiple myeloma. Cardiac amyloidosis was suspected based on the patient's age, the rapid development of severe heart failure, which did not respond well to symptomatic therapy, echocardiographic signs of restrictive heart disease (enlarged left atrium in the absence of left ventricular dilatation and decreased ejection fraction), symmetrical thickening of the interventricular septum and the wall of the left ventricle without outflow obstruction. tract, reducing the voltage of the QRS complex waves on the ECG. The latter feature distinguishes cardiac amyloidosis from LVH, although a true decrease in QRS amplitude (less than 5 mm in the limb leads and less than 10 mm in the chest leads) is observed only in half of patients with cardiac AL amyloidosis [14]. However, even in the absence of low voltage QRS complexes, its possible discrepancy with the degree of LVH on echocardiography should be taken into account. The results of cardiac MRI are of important diagnostic value, which allows not only to measure the mass of the left ventricular myocardium, but also to detect diffuse accumulation of gadolinium in the subendocardium [15].
In the presented observation, the diagnosis was confirmed by myocardial biopsy, although other tissues that are more accessible for biopsy can be used for histological examination, including the mucous membrane of the rectum or duodenum, subcutaneous fat, and kidney. The presence of AL amyloidosis was indicated by monoclonal secretion of lambda chains of immunoglobulins, detected using the Freelite method, as well as a decrease in the ratio of kappa and lambda chains to 0.01 (AL amyloidosis is characterized by a value of <0.26 or >1, 65). In 7-10% of patients, AL amyloidosis develops as part of multiple myeloma, to exclude which all patients should undergo a bone marrow biopsy.
This case demonstrates the possibility of “isolated” cardiac involvement in AL amyloidosis, although most patients have other manifestations, including proteinuria/nephrotic syndrome, enlarged liver and spleen, macroglossia, periorbital purpura, diarrhea, neuropathy, and/or orthostatic hypotension. The patient had slight proteinuria, but it could be due to blood stagnation in the systemic circulation.
Modern chemotherapy, including the proteasome inhibitor bortezomib, can achieve a complete or partial hematological response in a significant proportion of patients with AL amyloidosis, preventing amyloid deposition in other organs and the progression of heart failure.
P.P. Tao. Patient V., 65 years old, Russian, was examined at the clinic named after. EAT. Tareeva in December 2015. Over the course of three years, there has been an increasing decrease in pain, temperature and tactile sensitivity in the area of the hands and feet, like “gloves” and “socks”, and within one year – progressive congestive heart failure. Electromyography revealed gross axonal demyelinating disorders, most pronounced in the peroneal nerves, and echocardiography revealed a picture of restrictive heart damage: dilatation of the left atrium, thickening of the walls of the left ventricle, zones of hypokinesia in the interventricular septum, thickening of the endocardium, normal ejection fraction of the left ventricle. Brain natriuretic propeptide levels were elevated 30 times the upper limit of normal. Coronary angiography revealed stenosis of the anterior interventricular branch (65%) and the right coronary artery (75%). Percutaneous coronary angioplasty and stenting of the right coronary artery were performed, but after the intervention, heart failure persisted and orthostatic arterial hypotension appeared. According to echocardiography, the thickness of the interventricular septum reached 22 mm, multiple foci of granularity were detected in the myocardium, and the left ventricular ejection fraction decreased from 57% to 45%. MRI of the heart with gadolinium contrast against the background of pronounced thickening of the walls of the left ventricle revealed a circular subendocardial diffuse accumulation of the contrast agent in the myocardium of the left and anterior wall of the right ventricle (Fig. 5). A repeat biopsy of the rectal mucosa was performed, but amyloid could not be detected. An immunochemical study excluded monoclonal secretion of immunoglobulin light chains, characteristic of AL amyloidosis. A molecular genetic study revealed a mutation in the TTR gene (Val30Met), confirming the diagnosis of hereditary ATTR amyloidosis. Myocardial scintigraphy with 99mTcPYP revealed grade 2 accumulation of the radioisotope drug, and therefore it was decided to refrain from myocardial biopsy. In order to restore the tetrameric structure of mutant transthyretin, the patient receives tafamidis 20 mg/day for 2 years. The treatment was tolerable and there was no significant progression of amyloidosis.
Rice. 5. Symmetrical cardiac wall thickening and gadolinium accumulation in the subendocardium on MRI in a patient with ATTR amyloidosis
V.V. Rameev. As in the previous observation, amyloidosis was suspected in a 65-year-old patient due to a typical restrictive lesion of the left ventricle, characterized by the development of severe heart failure in the absence of dilatation and a significant decrease in the ejection fraction of the left ventricle. Echocardiography revealed multiple foci of granularity, which are often detected in patients with amyloid infiltration of the myocardium, and cardiac MRI revealed diffuse accumulation of gadolinium in the subendocardium. Systemic amyloidosis was also supported by peripheral polyneuropathy, which occurs in both AL and familial ATTR amyloidosis and may precede cardiac damage. In most cases, a steadily progressive, symmetrical distal neuropathy develops, starting with sensory disorders, primarily pain and temperature sensitivity, followed by disturbances in vibration and positional sensitivity and motor disorders. Early symptoms of neuropathy include paresthesia or painful dysesthesia. Carpal tunnel syndrome is often encountered, manifested by pain and paresthesia in the 1st-3rd fingers of the hand with gradual atrophy of the thenar muscles and caused by compression of the median nerve in the carpal tunnel by amyloid deposited in the wrist ligaments [16].
Considering the absence of monoclonal secretion of immunoglobulin light chains and the presence of a TTR gene mutation, a diagnosis of hereditary ATTRamyloidosis was made [17]. The diagnosis of amyloidosis should be confirmed by histological examination, but the results of a repeat rectal biopsy were negative. However, the presence of ATTRamyloidosis was not in doubt, taking into account the typical clinical picture and the results of molecular genetic studies. In addition, scintigraphy revealed grade 2 accumulation of 99mTcPYP in the myocardium (i.e., moderate accumulation, consistent with that in bone tissue). In 2021, a multicenter study showed that the accumulation of grade 2-3 99mTcPYP in the myocardium (Fig. 6) in the absence of monoclonal gammopathy has 100% specificity in diagnosing ATTR cardiac amyloidosis and actually eliminates the need for myocardial biopsy [18]. Moreover, scintigraphy with 99mTcPYP makes it possible to differentiate ATTR amyloidosis from AL amyloidosis, in which the accumulation of radioactive drug in the myocardium is absent or does not exceed grade 1.
Rice. 6. Accumulation of 99mTcPYP in the myocardium grade 3 in ATTR amyloidosis
Since the mid-90s of the 20th century, liver transplantation has been used to treat ATTRamyloidosis, which allows restoring the synthesis of normal trans thyretin. In recent years, drug stabilization of the tetrameric structure of transthyretin and prevention of the formation of amyloidogenic protein monomers have been considered a more promising treatment tactic. The first such drug, tafamidis, is already being used in Europe and the Russian Federation.
S.V. Moiseev. The presented observations illustrate a wide range of causes of LVH of unknown etiology, which include not only true myocardial hypertrophy, but also some infiltrative diseases, such as systemic amyloidosis, mimicking HCM. It is quite difficult to develop a clear algorithm for the differential diagnosis of HCM, given the variability of the course of diseases accompanied by LVH. For example, some severe genetic diseases, such as Pompe disease (type II glycogenosis associated with deficiency of the enzyme acid α-glucosidase in lysosomes), can appear at the age of 40-50 years and older, while in systemic diseases, including Fabry disease or amyloidosis, extracardiac symptoms are sometimes absent. A key role in the diagnosis of hereditary diseases, such as HCM, ATTRamyloidosis, Fabry disease, Pompe disease, Danon disease, etc., is played by the study of family history and molecular genetic research.
Myocardial hypertrophy: treatment
The goal of treating myocardial hypertrophy is to reduce the size of the heart ventricle. To solve this problem, an integrated approach is required, which is expressed in changing lifestyle and giving up bad habits: - exclude foods high in animal fats from your diet as much as possible; - stop smoking and drinking alcohol; — lead an active lifestyle (both at work and at leisure); — harden your body (baths and contrast showers are excellent remedies); Complex therapy using Transfer Factor Cardio is very effective - an immune drug based on transfer factor immune molecules, which, when entering the body, perform 3 functions: - eliminate violations in the structure of our DNA, which is the real cause of diseases; - strengthen our immune system against heart and vascular diseases; - act as carriers of “immune memory”, recording all the information about foreign elements that our body has had to deal with and methods for neutralizing them. These properties of this immunomodulator make it the most effective in the world, so its help is required not only for the complex treatment of myocardial hypertrophy, but also for any diseases of the heart and blood vessels. Myocardial hypertrophy, the treatment of which we discussed above, is actually not a “sentence.” With an appropriate lifestyle, any person can completely get rid of this disease and not remember about it, but taking an immunomodulator is simply necessary.
Symptoms
As a rule, this pathology is discovered accidentally, during an ECG or Echo-CG. Experts identify the following main signs of myocardial hypertrophy:
- angina pectoris;
- atrial fibrillation;
- high blood pressure;
- general weakness, poor health.
The development of angina occurs as a result of compression of the blood vessels that provide the necessary nutrition to the heart muscle. Ultimately, the muscle increases in size. It begins to consume more oxygen in combination with nutrients. Patients suffering from cardiac myocardial hypertrophy may also experience a condition during which the heart freezes for a few moments (does not beat at all). In this case, the person loses consciousness.
Get a free consultation Consultation does not oblige you to anything
Diagnostics
As a rule, the disease is detected by ultrasound. MRI of the heart is considered the simplest, most informative method. In certain cases, the disease is diagnosed using an ECG. Additional studies include: ventriculography, coronary angiography, and radioisotope studies.
Prevention
Effective preventive actions to prevent the development of the disease include:
- lifestyle changes: quitting smoking, drinking alcoholic beverages;
- combating risk factors: normalizing body weight, blood pressure;
- control of hyperlipidemia and hypertension with drugs (if lifestyle correction does not result).
If controlled with medications, it is possible to maintain normal sugar levels, as well as control other risk factors that occur with diabetes. Compliance with such preventive measures allows you to avoid the development of cardiac myocardial hypertrophy.
TYPES OF HYPERTROPHY OF HUMAN SKELETAL MUSCLES
Samsonova A.V. Types of hypertrophy of human skeletal muscles // Proceedings of the Department of Biomechanics of the University P.F. Lesgafta, 2021.- Issue. 14.- pp. 32-39
annotation
The article provides a classification of various types of human skeletal muscle hypertrophy based on a number of classification characteristics: time of manifestation and duration of preservation; direction of training impact; specific effects on myofibrils. Based on the time of manifestation and duration of maintenance of the training effect, short-term and long-term hypertrophy of skeletal muscles is distinguished. According to the direction of the training effect, myofibrillar and sarcoplasmic hypertrophy are distinguished. Based on the specific effects on myofibrils, it is proposed to distinguish between transverse and longitudinal myofibrillar hypertrophy.
Keywords
skeletal muscles, hypertrophy