Thalassemia is a genetic blood disease in which, due to gene mutations, an insufficient amount of hemoglobin is formed in the body and red blood cells are deformed.
Hemoglobin is a specific protein that is found in red blood cells and is responsible for the transport of iron and oxygen to all organs and tissues. The main function of hemoglobin is respiratory and its deficiency leads to oxygen starvation. As a result, organs cannot fully perform their physiological functions.
There are “adult” hemoglobin (HbA) and fetal hemoglobin (HbF).
Fetal (fetal) hemoglobin is synthesized in the fetus 2 weeks after the formation of internal organs (from approximately the 12th week of pregnancy). After the birth of a child, HbF is up to 80% and is gradually replaced by “adult” HbA.
Normally, in a person, the fetal type of hemoglobin should remain no more than 1.5%. If there is a shift of numbers to the left or to the right, this always indicates a pathology (the functioning of the cardiovascular system, thyroid gland, pituitary gland may be disrupted, the presence of hematological abnormalities, leukemia, lymphogranulomatosis, intoxication).
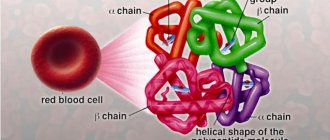
A healthy mature hemoglobin molecule (HbA) consists of two pairs of chains - alpha and beta. Fetal hemoglobin (HbF) consists of chains - gamma and delta.
And thalassemia is the result of impaired synthesis of one or more hemoglobin chains. On the basis of which alpha thalassemia, beta thalassemia, beta delta thalassemia, etc. are distinguished.
The severity of one or another type of disease depends on the number of damaged areas:
- If one gene of the chain is changed, the disease (thalassemia minor) is mild and asymptomatic. But, at the same time, the person remains a carrier of this disease, which he can pass on to his children.
- Violation of the synthesis of 2 genes is manifested by mild microcytic anemia.
- The pathology of 3 genes is severe with pronounced symptoms.
- Damage to 4 genes at once (thalassemia major) is the rarest type of thalassemia, which is practically incompatible with life (sometimes the situation is solved by a bone marrow transplant).
Alpha thalassemia major is the most serious disorder that occurs during fetal development. It threatens complications for both the child and the woman.
Today, with timely intrauterine transfusion of red blood cells, it is possible to save the life of the fetus.
Beta thalassemia major (Cooley's anemia) is also a dangerous disorder that manifests itself in the first two years of a child's life.
Alpha and beta thalassemia minor disrupt the process of erythropoiesis (the development of new red blood cells), which leads to chronic anemia and impaired hemolysis.
Regardless of the type, the clinical course of thalassemia is similar, the differences depend on the severity of the pathological process.
What is thalassemia and why does it occur?
Thalassemia is a genetic blood disorder. An insufficient amount of hemoglobin, the protein responsible for transporting oxygen and carbon dioxide, is formed. It consists of four peptides. 90% of hemoglobin molecules contain two alpha globins and two beta globins (hemoglobin A or A1), 2.5% contain delta chains instead of beta chains (hemoglobin A2), and the rest are hemoglobin A that has aged during use in red blood cells ( hemoglobin A3). When a mutation occurs in the genes responsible for the synthesis of one of the globins, the composition of hemoglobin is disrupted. These changes lead to the death of red blood cells.
What are the prospects for Zinteglo?
Since Zinteglo is conditionally approved (without strict requirements, as in the case of other drugs), Bluebird is required to annually send updated clinical data to the regulator so that it renews the registration of the drug, based on the fact that the benefits outweigh the harm. As soon as the data is collected into a pool of the required volume, it will be possible to talk about a full-fledged marketing permit.
In the future, patients with a genotype characterized by a complete absence or critically low endogenous synthesis of beta-globin will be added to the spectrum of patients for whom Zynteglo gene therapy is indicated: for example, β0/β0, mutations IVS-I-110 or IVS-I-5. The effectiveness of treatment in such patients may be less clear: clinical trials of HGB-212 are testing this.
This year, Bluebird intends to submit a registration dossier for Zinteglo to the US Food and Drug Administration (FDA), with a launch expected in 2020.
Along the way, Bluebird is completing a test of Zynteglo in the gene therapy treatment of sickle cell anemia, since this pathology is caused by other mutations of the same beta-globin gene.
The estimated European population of patients with transfusion-dependent beta thalassemia is 11–13 thousand people, with half living in Italy. Commercial prospects in the United States are much more modest with a patient pool of 1,400–1,500 people. Taking into account the upcoming inclusion of sickle cell anemia, it is quite possible for Zinteglo to earn bestseller status, crossing the threshold of $1 billion in annual sales.
In terms of competition, luspatercept will see the light of day in the foreseeable future: the FDA will decide on its approval for transfusion-dependent beta thalassemia by early December. Luspatercept, being developed by Acceleron Pharma and Celgene, is an erythrocyte maturation activator. This soluble fusion protein, comprising a modified extracellular domain of activin receptor type IIB (ActRIIB) and the Fc domain of human immunoglobulin IgG1, targets specific ligands of the transforming growth factor beta (TGF-β) superfamily that regulate late-stage erythropoiesis. Luspatercept is thought to act as a ligand decoy that inhibits abnormal Smad2/3 signaling responsible for ineffective erythropoiesis.
Causes and risk factors for developing the disease
Thalassemia is a hereditary disease, meaning the mutation is passed from parent to child. If one of your relatives suffered from this pathology, the risk of developing the disease increases.
The second risk factor is ethnicity. Thalassemia is most common in Africa, Central Asia and the Mediterranean countries, where it was discovered (translated from Greek “thalassemia” is “sea anemia”). There are two main types of thalassemia: alpha and beta. In the first case, the mutation affects the genes responsible for the synthesis of alpha globins, in the second - beta.
Alpha globins are encoded by four genes. The severity of the disease will depend on the number of pathologically altered DNA sections:
- 1 altered gene – asymptomatic form. But with it, a person becomes a carrier of the disease and can pass it on to his children;
- 2 genes – mild course of the disease;
- 3 genes – severe course of the disease;
- 4 genes is a rare type of disease that is poorly compatible with life. Most fetuses die during fetal development, and newborn babies generally die soon after birth or require lifelong therapy. In some cases, they can be cured by bone marrow transplantation.
Beta globins are encoded by a single gene, which is localized on chromosome 11. If the defective gene is contained in only one chromosome of a pair, the disease is mild (thalassemia minor). Damage to both chromosomes results in a very serious disease known as thalassemia major or Cooley's disease (Cooley's anemia).
Content
- 1 Signs and symptoms
- 2 Reason 2.1 Mutations
- 2.2 mRNA assembly
- 4.1 DNA analysis
- 6.1 Beta thalassemia major 6.1.1 Surgical
- 7.1 Evolutionary adaptation
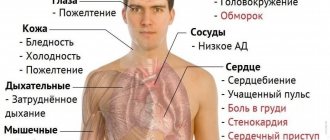
Symptoms and signs of thalassemia
In most cases, thalassemia is detected at the stage of prenatal diagnosis. If necessary, treatment begins immediately, without waiting for symptoms to appear. If the disease is not detected by prenatal diagnosis, the following symptoms are expected:
- pallor or yellowness of the mucous membranes;
- slow growth;
- dark urine;
- abdominal enlargement;
- deformation of bones, especially the bones of the skull.
The time at which the first signs of thalassemia appear largely depends on the type of disease and the number of mutations. In some children, symptoms are registered soon after birth, in others - in the first two years of life.
Diagnosis of thalassemia
Symptoms of thalassemia are more or less characteristic. To make a final diagnosis, the doctor needs laboratory results. If thalassemia is suspected, a general blood test is required. It will show a reduced number of red blood cells that are small, light, different in shape and size. In addition to mature cells, the smear will contain many of their precursors - blasts. Additionally, other specific blood tests may be prescribed to determine the severity of the disorders (biochemical analysis, determination of iron-binding capacity of plasma or serum ferritin). Molecular tests (PCR) have also been developed to determine the presence of mutations.
Ultrasound is used to assess the condition of the liver and spleen, and radiography is used to identify bone tissue pathology.
In a child, thalassemia can be diagnosed during pregnancy. This study is especially recommended for parents who are sick or may be carriers of this disease. There are two diagnostic methods:
- chorionic villus biopsy - performed at the 11th week of pregnancy;
- amniocentesis (sampling of amniotic fluid) - prescribed at the 16th week.
Links[edit]
- ^ abc "Beta thalassemia". A Home Guide to Genetics
. Retrieved May 26, 2015. - ^ a b c Advani, Puja. "Treatment and management of beta thalassemia". Medscape
. Retrieved April 4, 2021. - ^ a b McKinney, Emily Sloan; James, Susan R.; Murray, Sharon Smith; Nelson, Christina; Asheville, Jean (2014-04-17). Caring for mother and child. Elsevier Health Sciences. ISBN 9780323293778.
- Galanello, Renzo; Origa, Rafaella (21 May 2010). "Beta thalassemia". Orphanet J Rare Dis
.
5
: 11. DOI: 10.1186/1750-1172-5-11. PMC 2893117. PMID 20492708. - Goldman, Lee; Schafer, Andrew I. (April 21, 2015). Goldman-Cecil Medicine: Expert Consult - Online. Elsevier Health Sciences. ISBN 9780323322850.
- Carton, James (2012-02-16). Oxford Handbook of Clinical Pathology. OUP Oxford. ISBN 9780191629938.
- Perkin, Ronald M.; Newton, Dale A.; Swift, James D. (2008). Pediatric Hospital Medicine: A Textbook of Inpatient Care. Lippincott Williams and Wilkins. ISBN 9780781770323.
- Galanello, Renzo; Origa, Rafaella (21 May 2010). "Beta thalassemia". Journal "Orphanet of Rare Diseases"
.
5
(1): 11. DOI: 10.1186/1750-1172-5-11. ISSN 1750-1172. PMC 2893117. PMID 20492708. - Introduction to Pathology for the Physical Therapist Assistant. Jones and Bartlett Publishing. 2011. ISBN. 9780763799083.
- Anderson, Gregory J.; McLaren, Gordon D. (January 16, 2012). Iron Physiology and Human Pathophysiology. Springer Science & Business Media. ISBN 9781603274845.
- Barton, James S.; Edwards, Corwin Q.; Phatak, Pradyumna D.; Britton, Robert S.; Bacon, Bruce R. (2010-07-22). Handbook of Iron Overload Disorders. Cambridge University Press. ISBN 9781139489393.
- McCance, Catherine L.; Hueter, Sue E. (December 13, 2013). Pathophysiology: the biological basis of disease in adults and children. Elsevier Health Sciences. ISBN 9780323088541.
- Leonard, Debra GB (2007-11-25). Molecular pathology in clinical practice. Springer Science & Business Media. ISBN 9780387332277.
- Bowen, Juan M.; Mazzaferri, Ernest L. (2012-12-06). Modern Internal Medicine: Clinical Cases. Springer Science & Business Media. ISBN 9781461567134.
- Disorders, National Rare Organization (2003). NORD Guide to Rare Diseases. Lippincott Williams and Wilkins. ISBN 9780781730631.
- Barton, James S.; Edwards, Corwin K. (January 13, 2000). Hemochromatosis: genetics, pathophysiology, diagnosis and treatment. Cambridge University Press. ISBN 9780521593809.
- Wilkins, Lippincott Williams & (2009). Professional reference book on diseases. Lippincott Williams and Wilkins. paragraph 513. ISBN 9780781778992. thalassemia major.
- Ward, Amanda J; Cooper, Thomas A (2009). "The pathobiology of fusion". Journal of Pathology
.
220
(2):152–63. DOI: 10.1002/path.2649. PMC 2855871. PMID 19918805. - "DNA determination". Dictionary.com
. Retrieved May 26, 2015. - Okpala, Iheanyi (2008-04-15). Practical management of hemoglobinopathies. John Wiley and Sons. ISBN 9781405140201.
- Vasudevan, D.M.; Sreekumari, S.; Vaidyanathan, Kannan (11/01/2011). Textbook of Biochemistry for Dental Students. JP Medical Ltd. ISBN 9789350254882.
- Taeusch, H. William; Ballard, Roberta A.; Gleason, Christine A.; Avery, Mary Ellen (2005). Avery's diseases of the newborn. Elsevier Health Sciences. ISBN 978-0721693477.
- Beta Thalassemia: New Insights for the Healthcare Professional: 2013 Edition: ScholarlyBrief. Scientific publications. 2013-07-22. ISBN 9781481663472.
- "Risk factors" . Mayo Clinic
. Retrieved April 4, 2021. - “How are thalassemias diagnosed? - NHLBI, NIH." www.nhlbi.nih.gov
. Retrieved May 26, 2015. - Target Cells, Imperial College London School of Medicine
- ^ ab Orkin, Stewart H.; Nathan, David J.; Ginsburg, David; See, A. Thomas; Fisher, David E.; Lux, Samuel (2009). Nathan and Oska's Hematology of Infancy and Childhood
(7th ed.).
Philadelphia: Saunders. ISBN 978-1-4160-3430-8.[ page required
] - “What are the signs and symptoms of thalassemia? - NHLBI, NIH." www.nhlbi.nih.gov
. Retrieved May 26, 2015. - Galanello, Renzo; Origa, Rafaella (2010). "Beta thalassemia". Journal "Orphanet of Rare Diseases"
.
5
(1): 11. DOI: 10.1186/1750-1172-5-11. PMC 2893117. PMID 20492708. - Jump up
↑ Schrijver, Iris (09/09/2011). Diagnostic molecular pathology in practice: an individual approach. Springer Science & Business Media. ISBN 9783642196775. - Cousins, NE; Gaff, C. L.; Metcalfe, S. A.; Delatitsky, M. B. (2010). "Screening for carriers of beta thalassemia: a review of international practice". European Journal of Human Genetics
.
18
(10): 1077–83. DOI: 10.1038/ejhg.2010.90. PMC 2987452. PMID 20571509. - "Screening for beta thalassemia trait: dangers in populations of West African descent". Retrieved April 4, 2021.
- Muncy, Herbert L.; Campbell, James S. (2009). "Alpha and beta thalassemia". American family physician
.
80
(4): 339–44. PMID 19678601. - Greer, John P.; Arber, Daniel A.; Glader, Bertil; List, Alan F.; So Robert T.; Paraskevas, Frixos; Rogers, George M. (August 29, 2013). Wintrobe Clinical Hematology. Lippincott Williams and Wilkins. ISBN 9781469846224.
- Greer, John P.; Arber, Daniel A.; Glader, Bertil; List, Alan F.; So Robert T.; Paraskevas, Frixos; Rogers, George M. (August 29, 2013). Wintrobe Clinical Hematology. Lippincott Williams and Wilkins. ISBN 9781469846224.
- Hydroxamic Acids: Advances in Research and Application: 2011 Edition: ScholarlyPaper. Scientific publications. 2012-01-09. ISBN 9781464952081.
- "NCBI - WWW Error Blocking Diagnostics". pubchem.ncbi.nlm.nih.gov
. Retrieved May 26, 2015. - "Deferoxamine". ivertox.nih.gov
. Retrieved May 26, 2015. - Sabloff, Mitchell; Chandi, Mammen; Wang, Zhiwei; Logan, Brent R.; Gavamzade, Ardeshir; Lee, Chi-Kong; Irfan, Syed Mohammad; Bredeson, Christopher N.; Cowan, Morton J. (2011). "HLA-matched sibling bone marrow transplantation for β-thalassemia major". Blood
.
117
(5):1745–1750. DOI: 10.1182/blood-2010-09-306829. ISSN 0006-4971. PMC 3056598. PMID 21119108. - "Gene therapy shows promise for treating beta thalassemia and sickle cell disease". 2012-03-28. Retrieved October 15, 2015.
- Uranius, Selman. "Splenectomy for hematological disorders". NCBI
. Retrieved April 4, 2021. - Ah, Cohen. "Blood transfusion for β-thalassemia major". NCBI
. Retrieved April 4, 2021. - CRISPR Therapeutics and Vertex Pharmaceuticals are making arrangements to begin the first CRISPR/Cas9 clinical trial in Europe in 2018. Clara Rodriguez Fernandez December 13, 2021
- Jump up
↑ Cappellini, Maria Domenica (2007).
"Exjade® (deferasirox, ICL670) in the treatment of transfusion-associated chronic iron overload". Therapy and clinical risk management
.
3
(2): 291–299. DOI: 10.2147/tcrm.2007.3.2.291. ISSN 1176-6336. PMC 1936310. PMID 18360637. - Advani, Puja. "A cure for beta thalassemia". Medscape
. Retrieved April 4, 2021. - Schwartz, M. William (2012). Pediatric consultation in 5 minutes. Lippincott Williams and Wilkins. ISBN 9781451116564.
- Porvit, Anna; McCullough, Jeffrey; Erber, Wendy N. (May 27, 2011). Pathology of blood and bone marrow. Elsevier Health Sciences. ISBN 978-0702045356.
- Hemoglobinopathies. Published by Jaypee Brothers. 2006. ISBN 9788180616693.
- Torre, Dario M.; Lamb, Jeffrey S.; Ruisvik, Jerome Van; Shapira, Ralph M. (2009). Kochar "Clinical medicine for students". Lippincott Williams and Wilkins. ISBN 9780781766999.
- Brissot, Pierre; Cappellini, Maria Domenica (2014). "LIVER DISEASE". International Thalassemia Federation. Quote journal requires |journal=(help)
- “WHO | Global epidemiology of hemoglobin disorders and derived maintenance indicators". www.who.int
. Retrieved May 26, 2015. - Berg, Sheri; Bittner, Edward A. (2013-10-16). MGH Critical Care Medicine Review. Lippincott Williams and Wilkins. ISBN 9781451173680.
- Hematology made simpler. AuthorHouse. 2013-02-06. ISBN 9781477246511.
- Abouelmagd, Ahmed; Ageli, Hussein M. (2013). Fundamentals of Genetics: Primer covering the molecular composition of genetic material, gene expression and genetic engineering, and mutations and human genetics. Universal Publishers. ISBN 9781612331928.
- Weatherall, David J (2010). "Chapter 47. Thalassemias: disorders of globin synthesis". In Lichtman, Massachusetts; Kipps, T. J.; Seligsohn, U; Kaushansky, K; Prchal, J. T. (ed.). Thalassemias: disorders of globin synthesis
.
Williams Hematology
(8th ed.). McGraw-Hill Companies. - "Statistics on Thalassemia". Correct diagnosis
. Retrieved April 4, 2021. - "Thalassemia: Genetic blood disorder expected to double in next few decades". ScienceDaily
. Retrieved April 4, 2021.
Treatment of thalassemia
Determined by type and severity. For moderately severe symptoms, treatment is not prescribed. From time to time only blood transfusions are performed. This is mainly necessary after operations, childbirth, or to prevent possible complications. People with beta thalassemia require more frequent blood transfusions. To normalize excess iron levels, they are also prescribed specific drugs that bind and remove iron.
For severe and severe forms of the disease, there are two treatment options:
- frequent blood transfusions (every few weeks), which are combined with taking medications that remove excess iron from the body;
- Bone marrow transplant is the only method that can completely cure a person of thalassemia. Unfortunately, transplantation is not always successful.
Medicines for thalassemia are prescribed only to correct symptoms and complications. There is no drug therapy for the disease itself.
How much does Zinteglo cost?
PS Bluebird put the cost of Zinteglo at 1.575 million euros ($1.77 million). As expected, payment will be made in installments over five years in equal installments: first an advance, and then annual payments, but only if the effectiveness of treatment is recorded.
Bluebird has not yet determined the final cost of its gene therapy, but it will obviously be prohibitive. It is clear that in highly developed countries with medical insurance, none of the patients pays the entire amount out of their own pocket.
Yes, according to Bluebird's calculations, the "fair price" of Zinteglo, calculated taking into account the improvement in the patient's quality and life expectancy, is within $2.1 million, but the final cost is expected to be significantly less. This astronomical amount obviously comes from the total costs of the health care system for many years of chronic therapy for beta thalassemia. Thus, according to experts, one American patient with this disease costs somewhere between 200-300 thousand dollars annually.
It is proposed to pay 20% in advance in the first year, and the remaining amount to be paid in 20% installments annually for four years, provided that the effectiveness of treatment meets predetermined criteria, such as the disappearance of the need for blood transfusions or a significant reduction in the number of such procedures.
In other words, you will have to pay only based on the effectiveness of gene therapy, especially since there is still no indisputable data on the duration of its healing effect. The approach seems reasonable given the differences in intermediate outcomes among patients, although the overall effectiveness of Zynteglo is impressive.
According to industry experts, the cost of Zinteglo will be $1.2 million in the United States and $900 thousand (785 thousand euros) in Europe.
Complications of thalassemia
Possible complications of the disease:
- excess iron, which is part of hemoglobin. The accumulation of this element in the body leads to damage to the heart, liver, and endocrine system;
- susceptibility to infections. This is especially true for patients who have had their spleen removed;
- bone deformation associated with an increase in bone marrow volume. Most often, this process affects the bones of the skull, less often the limbs. They become thinner and break more often;
- splenomegaly is an enlargement of the spleen, where defective red blood cells mainly die. If the spleen is very enlarged, it is removed. This operation is called splenectomy;
- delayed growth and puberty;
- Heart diseases (chronic heart failure and arrhythmias) can develop in severe cases of the disease.
Further reading[edit]
- Cao, Antonio; Galanello, Renzo (2010). "Beta thalassemia". In Pagona, Roberta A; Bird, Thomas D; Dolan, Cynthia R.; Stevens, Karen; Adam, Margaret P. (ed.). GeneReviews. University of Washington, Seattle. PMID 20301599.
- Bahal, Raman; McNer, Nicole Ali; Quijano, Elias; Liu, Yanfeng; Sulkowski, Parker; Turchik, Audrey; Lu, Yi-Jian; Bhunia, Dinesh C.; Manna, Arunava; Greiner, Dale L.; Brehm, Michael A.; Cheng, Christopher J.; Lopez-Giraldez, Francesc; Ricciardi, Adele; Beloor, Jagadish; Krause, Diane S.; Kumar, Preeti; Gallagher, Patrick J.; Braddock, Demetrios T.; Salzman, W. Mark; Ly, Danith H.; Glaser, Peter M. (October 26, 2021). "In vivo correction of anemia in β-thalassemic mice using nanoparticle-delivered γPNA-mediated gene editing". Nature Communications
.
7
: 13304. Bibcode: 2016NatCo... 713304B. DOI: 10.1038/ncomms13304. ISSN 2041-1723. PMC 5095181. PMID 27782131.
Homozygote gives a big disease
Symptoms of beta thalassemia are determined by an imbalance of production
and
a change in the number of beta chains of the protein part of hemoglobin
, which mainly predisposes to ineffective hematopoiesis. Red blood cells that have not left the nuclear stage die en masse at the place of their birth - in the bone marrow; adult blood cells (and to a lesser extent reticulocytes) prematurely end their lives in the spleen, as a result of which symptoms of severe anemia are formed. In addition, new sources of hematopoiesis are formed in the liver and spleen. High-intensity hematopoiesis occurring in the skeletal system disrupts its development (distortions and deformations), and the state of severe hypoxia entails a delay in the overall development of the child’s body. This variant of the course is predominantly present in homozygous thalassemia rather than in the heterozygous form.
Symptoms due to biparental involvement
Thalassemia major manifests itself already from the birth of a small person, however, in the first months the severity of the symptoms is not so obvious, but closer to the age of one year, in extreme cases - at the beginning of the second year of life, the clinical manifestations in children become quite bright:
- It is impossible not to notice the unnatural shape of the skull (a tower, one might say, square head), the large size of the upper jaw and the peculiar features of the child’s face: Mongoloid type, small eyes, a noticeably flattened bridge of the nose (R-graphy shows pathological changes);
- The pallor of the skin, the change in its color - from grayish to icteric - attracts attention;
- In children, the liver and spleen enlarge early, which is due to the extramedullary (extramedullary) type of hematopoiesis and uncontrolled deposition of hemosiderin in the tissues (it is formed during the breakdown of red blood cells);
- Children often get sick because their immune defenses are reduced, they are delayed in physical development, and children who are fortunate enough to have a milder form of homozygous beta thalassemia and are destined to live into adolescence and adulthood have delayed development of secondary sexual characteristics;
- Laboratory monitoring, mandatory in such cases, shows compliance with the clinical manifestations of severe hemolytic anemia.
Analyzes
Due to the fact that the main diagnostic criterion for thalassemia is laboratory tests, it is necessary to give a brief description of the blood picture that develops in this pathology:
- Hemoglobin level is reduced to 50-30 g/l;
- The color indicator in calculations shows a disappointing result - 0.5 and below;
- The erythrocytes in the smear are hypochromic, the cell sizes are changed (pronounced anisocytosis), target-like appearance and basophilic granularity characteristic of beta-thalassemia (and membranopathy) are noted;
- The osmotic resistance of red blood cells is increased;
- The number of reticulocytes is increased;
- Biochemical blood parameters (bilirubin due to the free fraction, serum iron) are increased. Excessive iron accumulation contributes to the formation of liver cirrhosis, leads to the development of diabetes mellitus and damage to the heart muscle.
A sign confirming the diagnosis in the patient’s blood tests is an increased content of fetal hemoglobin in red blood cells (up to 20-90%).
Treatment
Severe oxygen starvation of tissues and intensive work of the hematopoietic system in the bones (where a healthy person does not have it) are indications for the transfusion of full-fledged donor red blood cells capable of ensuring tissue respiration. In patients with thalassemia major, blood transfusions from infancy are the main therapeutic method of influencing the disease. At the same time, it should be understood that the transfusion of someone else’s blood does not belong to conventional medicines (tablets and solutions), so such treatment must be approached with extreme caution. Nevertheless:
- At the first stage, the patient is provided with a shock course of blood transfusions (within 2-3 weeks the patient receives up to 10 transfusions, which allows increasing the hemoglobin level to 120-140 g/l);
- At the next stage, the number of blood transfusions is reduced, but they try to keep Hb within 90-100 g/l.
This treatment tactic makes it possible to:
- Significantly improve your well-being;
- Reduce the negative impact of pathology on the skeletal system;
- Increase immunity;
- Prevent further enlargement of the spleen;
- It has a beneficial effect on the physical development of the baby.
However, despite the fact that whole blood transfusion has completely disappeared (in such cases it was replaced by red blood cells), the “non-native” biological environment still retains the risk of complications:
- Pyrogenic reactions (they occur mainly due to a violation of the technique of washing red blood cells);
- Excessive deposition of hemosiderin (iron-containing pigment), which contributes to liver enlargement, the development of cardiac pathology and diabetes.
In order to ensure the removal of excess iron from the body, the patient is prescribed Desferal in the form of intramuscular injections (the dose of the drug corresponds to the age and volume of transfused Ermass). The combination of Desferal with vitamin C (ascorbic acid) is quite useful in such situations.
A significant enlargement of the spleen, as well as accompanying changes in the blood (leukopenia, thrombocytopenia), provide grounds for removal of the constantly “growing” organ (splenectomy).
Meanwhile, stable long-term improvement in the condition of patients with homozygous β-thalassemia can be achieved with the help of bone marrow transplantation, but this operation presents certain difficulties in terms of selecting a donor-recipient pair and is fraught with the development of reactions after transplantation.
Finally, a few words about reducing the production of α-chains
The production of α-chains of red blood pigment is controlled by two pairs of genes, which makes possible the formation of different forms of the disease:
- If alpha chains are completely absent, then in the embryonic period of the child’s development, red blood pigment is not formed at all. In such cases, there is only one outcome - dropsy of the brain and the death of the child;
- If the functional abilities of 1 or 2 genes from two pairs are impaired, then the symptoms of the disease begin to very much resemble the clinical manifestations of β-thalassemia, although laboratory diagnostic indicators in terms of the presence of HbF and HbA2 vary significantly, the level of these hemoglobins does not tend to increase.
Therapy for α-thalassemia is, in general, no different from the treatments used to treat the heterozygous variant of β-thalassemia.
Care
Treatment for alpha thalassemia may include blood transfusions to maintain hemoglobin at levels that reduce symptoms of anemia. The decision to initiate transfusion depends on the clinical severity of the disease. [19] Splenectomy is a possible treatment option to increase total hemoglobin levels in cases of worsening anemia due to an overactive or enlarged spleen, or when transfusion therapy is not possible. [20] However, splenectomy should be avoided when other options are available due to the increased risk of serious infections and thrombosis. [20]
Additionally, gallstones may be a problem requiring surgery. Secondary complications following a fever attack should be monitored, and most people live without any treatment. [14]
Additionally, stem cell transplantation should be considered a treatment (and cure) that is best done early in life. Other options, such as gene therapy, are still being developed. [21]
A study by Kreger et al, combining a retrospective review of three cases of alpha thalassemia major and a literature review of 17 cases, showed that intrauterine blood transfusion can lead to favorable outcomes. Ultimately, successful hematopoietic cell transplantation was performed in four patients. [22]