Талассемия – это генетическая болезнь крови, при которой, из-за мутации генов, образовывается недостаточное количество гемоглобина в организме и происходит деформация эритроцитов.
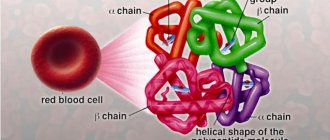
Гемоглобин – это специфический белок, который содержится в эритроцитах и отвечает за перенос железа и кислорода ко всем органам и тканям. Основная функция гемоглобина – дыхательная и его недостаток приводит к кислородному голоданию. В результате чего органы не могут полноценно выполнять свои физиологические функции.
Различают «взрослый» гемоглобин (HbA) и плодный тип гемоглобина (HbF).
Фетальный (плодный) гемоглобин синтезируется у плода спустя 2 недели после формирования внутренних органов (приблизительно с 12-й недели беременности). После рождения ребенка, HbF составляет до 80% и постепенно замещается «взрослым» HbA.
В норме у человека, плодного типа гемоглобина должно остаться не более 1,5%. Если случился сдвиг цифр влево или вправо – это всегда говорит о патологии (может быть нарушена работа сердечно-сосудистой системы, щитовидной железы, гипофиза, наличие гематологических отклонений, лейкоза, лимфогранулематоза, интоксикации).
Здоровая зрелая молекула гемоглобина (HbA) состоит из двух пар цепей – альфы и беты. Фетальный гемоглобин (HbF) состоит из цепей – гаммы и дельты.
А талассемия – это результат нарушенного синтеза одной или нескольких цепей гемоглобина. На основе чего различают альфа-талассемию, бета-талассемию, бета-дельту талассемию и т.д.
Тяжесть протекания того или другого вида заболевания зависит от количества поврежденных участков:
- Если изменен один ген цепи – заболевание (малая талассемия), протекает легко и бессимптомно. Но, при этом человек остается носителем данной болезни, которую может передать по наследству своим детям.
- Нарушение синтеза 2-х генов проявляется микроцитарной анемией в легкой степени тяжести.
- Патология 3-х генов протекает тяжело с ярко выраженной симптоматикой.
- Повреждение сразу 4-х генов (большая талассемия) – это наиболее редкий вид талассемии, который практически несовместим с жизнью (иногда ситуация решается пересадкой костного мозга).
Большая альфа-талассемия – самое серьезное отклонение, которое возникает еще в период внутриутробного развития плода. Грозит осложнениями, как для ребенка, так и для женщины.
Сегодня при своевременной внутриутробной трансфузии эритроцитарной массы удается сохранить жизнь плода.
Большая бета-талассемия (анемия Кули) – это также опасное нарушение, которое проявляется в первые два года жизни ребенка.
Малая альфа и бета-талассемия нарушает процесс эритропоэза (развитие новых эритроцитов), что приводит к хронической анемии и нарушению гемолиза.
Вне зависимости от вида, клиническое протекание талассемий схожи, различия зависят от степени тяжести патологического процесса.
Что такое талассемия и почему она возникает
Талассемия – это генетическое заболевание крови. Образуется недостаточное количество гемоглобина, белка, отвечающего за перенос кислорода и углекислого газа. Он состоит из четырех пептидов. 90% молекул гемоглобина содержат два альфа-глобина и два бета-глобина (гемоглобин А или A1), 2.5% содержат вместо бета-цепей дельта-цепи (гемоглобин А2), а остальные представляют собой гемоглобин A, состарившийся в процессе эксплуатации в эритроцитах (гемоглобин A3). При мутации в генах, отвечающих за синтез одного из глобинов, состав гемоглобина нарушается. Эти изменения влекут гибель эритроцитов.
Какие перспективы у «Зинтегло»?
Поскольку «Зинтегло» одобрен условно (без жёстких требований, как в случае других лекарственных средств), «Блюбёрд» обязана ежегодно отправлять регулятору обновленные клинические данные, чтобы тот продлевал регистрацию препарата, исходя из факта перевешивания пользы над вредом. Как только данные соберутся в пул должного объема, можно будет говорить о полноценном маркетинговом разрешении.
В дальнейшем к спектру пациентов, которым показана генотерапия «Зинтегло», будут добавлены больные с генотипом, характеризующимся полным отсутствием или критически малым эндогенным синтезом бета-глобина: например, β0/β0, мутации IVS-I-110 или IVS-I-5. Возможно, эффективность лечения таких пациентов менее выражена: проверка осуществляется в клинических испытаниях HGB-212.
В текущем году «Блюбёрд» намеревается отправить в Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) регистрационное досье на «Зинтегло», запуск ожидается в 2020 году.
Попутно «Блюбёрд» завершает проверку «Зинтегло» в генно-терапевтическом лечении серповидно-клеточной анемии, поскольку эта патология вызвана иными мутациями того же гена бета-глобина.
Оценочно европейская популяция больных зависимой от гемотрансфузий бета-талассемией составляет 11–13 тыс. человек, причем половина проживает в Италии. Коммерческие перспективы в Соединенных Штатах гораздо скромнее с их пациентским пулом в пределах 1,4–1,5 тыс. человек. С учетом грядущего подключения серповидно-клеточной анемии для «Зинтегло» вполне реально заработать статус бестселлера, перешагнув порог в 1 млрд долларов годовых продаж.
Что касается конкуренции, в обозримом будущем свет увидит луспатерцепт (luspatercept): до начала декабря FDA примет решение относительно его одобрения в терапии бета-талассемии, зависимой от гемотрансфузий. Луспатерцепт, разрабатываемый «Акселерон фарма» (Acceleron Pharma) и «Селджен» (Celgene), представляет собой активатор созревания эритроцитов. Этот растворимый гибридный белок, включающий модифицированный внеклеточный домен активинового рецептора типа IIB (ActRIIB) и Fc-домен иммуноглобулина человека IgG1, таргетирован на определенные лиганды суперсемейства трансформирующего фактора роста бета (TGF-β), регулирующие эритропоэз на поздней стадии. Считается, что луспатерцепт работает как лигандная ловушка, сдерживающая патологическую сигнализацию Smad2/3, ответственную за неэффективный эритропоэз.
Причины и факторы риска развития заболевания
Талассемия – заболевание наследственное, то есть мутация передается от родителя ребенку. Если кто-то из родственников страдал от этой патологии, риск развития заболевания повышается.
Второй фактор риска – этническая принадлежность. Больше всего талассемия распространена в Африке, Средней Азии и странах Средиземноморья, где она и была открыта (в переводе с греческого “талассемия” — это “морская анемия”). Различают два основных типа талассемии: альфа и бета. В первом случае мутация затрагивает гены, отвечающие за синтез альфа-глобинов, во втором – бета.
Альфа-глобины кодируются четырьмя генами. Тяжесть заболевания будет зависеть от количества патологически измененных участков ДНК:
- 1 измененный ген – бессимптомная форма. Но при ней человек становится носителем заболевания и может передать его своим детям;
- 2 гена – легкое течение болезни;
- 3 гена – тяжелое течение болезни;
- 4 гена – редкий тип заболевания, который плохо совместим с жизнью. Большинство плодов гибнет еще в период внутриутробного развития, а родившиеся малыши, в основном, умирают вскоре после рождения или нуждаются в пожизненной терапии. В отдельных случаях удается их вылечить путем пересадки костного мозга.
Бета-глобины кодируются одним геном, который локализуется в 11-й хромосоме. Если дефектный ген содержится только в одной хромосоме из пары, заболевание протекает легко (малая талассемия). Повреждение обеих хромосом приводит к очень серьезному заболеванию, известному как большая талассемия или болезнь Кули (анемия Кули).
Содержание
- 1 Признаки и симптомы
- 2 Причина 2.1 Мутации
- 2.2 сборка мРНК
- 4.1 Анализ ДНК
- 6.1 Большая бета-талассемия 6.1.1 Хирургический
- 7.1 Эволюционная адаптация
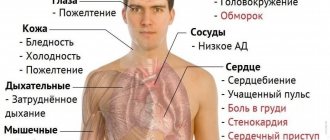
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников — бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Ссылки [ править ]
- ^ a b c «Бета-талассемия» . Домашний справочник по генетике
. Проверено 26 мая 2015 . - ^ а б в Адвани, Пуджа. «Лечение и лечение бета-талассемии» . Medscape
. Проверено 4 апреля 2021 года . - ^ а б МакКинни, Эмили Слоун; Джеймс, Сьюзан Р .; Мюррей, Шэрон Смит; Нельсон, Кристина; Эшвилл, Джин (2014-04-17). Уход за матерью и ребенком . Elsevier Health Sciences. ISBN 9780323293778.
- Галанелло, Ренцо; Орига, Рафаэлла (21 мая 2010 г.). «Бета-талассемия» . Orphanet J Rare Dis
.
5
: 11. DOI : 10,1186 / 1750-1172-5-11 . PMC 2893117 . PMID 20492708 . - Голдман, Ли; Шафер, Эндрю И. (21 апреля 2015 г.). Goldman-Cecil Medicine: Expert Consult — Online . Elsevier Health Sciences. ISBN 9780323322850.
- Картон, Джеймс (2012-02-16). Оксфордский справочник по клинической патологии . ОУП Оксфорд. ISBN 9780191629938.
- Перкин, Рональд М .; Newton, Dale A .; Свифт, Джеймс Д. (2008). Детская госпитальная медицина: Учебник стационарного лечения . Липпинкотт Уильямс и Уилкинс. ISBN 9780781770323.
- Галанелло, Ренцо; Орига, Рафаэлла (21 мая 2010 г.). «Бета-талассемия» . Журнал «Орфанет редких болезней»
.
5
(1): 11. DOI : 10,1186 / 1750-1172-5-11 . ISSN 1750-1172 . PMC 2893117 . PMID 20492708 . - Введение в патологию для ассистента физиотерапевта . Издательство «Джонс и Бартлетт». 2011. ISBN. 9780763799083.
- Андерсон, Грегори Дж .; Макларен, Гордон Д. (16 января 2012 г.). Физиология железа и патофизиология человека . Springer Science & Business Media. ISBN 9781603274845.
- Бартон, Джеймс С .; Эдвардс, Корвин Q .; Phatak, Pradyumna D .; Бриттон, Роберт С .; Бэкон, Брюс Р. (22.07.2010). Справочник по расстройствам, связанным с перегрузкой железом . Издательство Кембриджского университета. ISBN 9781139489393.
- Маккэнс, Кэтрин Л .; Хютер, Сью Э. (13 декабря 2013 г.). Патофизиология: биологическая основа болезней у взрослых и детей . Elsevier Health Sciences. ISBN 9780323088541.
- Леонард, Дебра GB (2007-11-25). Молекулярная патология в клинической практике . Springer Science & Business Media. ISBN 9780387332277.
- Боуэн, Хуан М .; Маззаферри, Эрнест Л. (2012-12-06). Современная внутренняя медицина: клинические примеры . Springer Science & Business Media. ISBN 9781461567134.
- Расстройства, Национальная организация редких (2003). Руководство NORD по редким заболеваниям . Липпинкотт Уильямс и Уилкинс. ISBN 9780781730631.
- Бартон, Джеймс С .; Эдвардс, Корвин К. (13 января 2000 г.). Гемохроматоз: генетика, патофизиология, диагностика и лечение . Издательство Кембриджского университета. ISBN 9780521593809.
- Wilkins, Липпинкотты Williams & (2009). Профессиональный справочник по болезням . Липпинкотт Уильямс и Уилкинс. п. 513 . ISBN 9780781778992. большая талассемия.
- Уорд, Аманда J; Купер, Томас А (2009). «Патобиология сращивания» . Журнал патологии
.
220
(2): 152–63. DOI : 10.1002 / path.2649 . PMC 2855871 . PMID 19918805 . - «определение ДНК» . Dictionary.com
. Проверено 26 мая 2015 . - Okpala, Iheanyi (2008-04-15). Практическое ведение гемоглобинопатий . Джон Вили и сыновья. ISBN 9781405140201.
- Васудеван, DM; Sreekumari, S .; Вайдьянатан, Каннан (01.11.2011). Учебник биохимии для студентов-стоматологов . JP Medical Ltd. ISBN 9789350254882.
- Taeusch, Х. Уильям; Ballard, Roberta A .; Глисон, Кристин А .; Эйвери, Мэри Эллен (2005). Болезни Эйвери новорожденных . Elsevier Health Sciences. ISBN 978-0721693477.
- Бета — талассемия: New Insights для Healthcare Professional: 2013 Издание: ScholarlyBrief . Научные издания. 2013-07-22. ISBN 9781481663472.
- «Факторы риска» . Клиника Мэйо
. Проверено 4 апреля 2021 года . - «Как диагностируются талассемии? — NHLBI, NIH» . www.nhlbi.nih.gov
. Проверено 26 мая 2015 . - Клетки-мишени , Имперский колледж Лондонского медицинского факультета
- ^ a b Оркин, Стюарт Х .; Натан, Дэвид Дж .; Гинзбург, Дэвид; Смотри, А. Томас; Фишер, Дэвид Э .; Люкс, Сэмюэл (2009). Гематология младенчества и детства Натана и Оски
(7-е изд.). Филадельфия: Сондерс. ISBN 978-1-4160-3430-8.[
требуется страница
] - «Каковы признаки и симптомы талассемии? — NHLBI, NIH» . www.nhlbi.nih.gov
. Проверено 26 мая 2015 . - Галанелло, Ренцо; Орига, Рафаэлла (2010). «Бета-талассемия» . Журнал «Орфанет редких болезней»
.
5
(1): 11. DOI : 10,1186 / 1750-1172-5-11 . PMC 2893117 . PMID 20492708 . - Перейти
↑ Schrijver, Iris (09.09.2011). Диагностическая молекулярная патология на практике: индивидуальный подход . Springer Science & Business Media. ISBN 9783642196775. - Cousens, NE; Gaff, CL; Metcalfe, SA; Делатицкий, МБ (2010). «Скрининг носителей бета-талассемии: обзор международной практики» . Европейский журнал генетики человека
.
18
(10): 1077–83. DOI : 10.1038 / ejhg.2010.90 . PMC 2987452 . PMID 20571509 . - «Скрининг на признак бета-талассемии: опасности среди населения западноафриканского происхождения» . Проверено 4 апреля 2021 года .
- Манси, Герберт Л .; Кэмпбелл, Джеймс С. (2009). «Альфа и бета талассемия» . Американский семейный врач
.
80
(4): 339–44. PMID 19678601 . - Грир, Джон П .; Arber, Daniel A .; Глэдер, Бертил; Лист, Алан Ф .; Значит, Роберт Т .; Параскевас, Фриксос; Роджерс, Джордж М. (29 августа 2013 г.). Клиническая гематология Винтроба . Липпинкотт Уильямс и Уилкинс. ISBN 9781469846224.
- Грир, Джон П .; Arber, Daniel A .; Глэдер, Бертил; Лист, Алан Ф .; Значит, Роберт Т .; Параскевас, Фриксос; Роджерс, Джордж М. (29 августа 2013 г.). Клиническая гематология Винтроба . Липпинкотт Уильямс и Уилкинс. ISBN 9781469846224.
- Гидроксамовые кислоты: достижения в исследованиях и применении: издание 2011 года: ScholarlyPaper . Научные издания. 2012-01-09. ISBN 9781464952081.
- «NCBI — Диагностика блокировки ошибок WWW» . pubchem.ncbi.nlm.nih.gov
. Проверено 26 мая 2015 . - «Дефероксамин» . ivertox.nih.gov
. Проверено 26 мая 2015 . - Саблофф, Митчелл; Чанди, Маммен; Ван, Чживэй; Логан, Брент Р .; Гавамзаде, Ардешир; Ли, Чи-Конг; Ирфан, Сайед Мохаммад; Bredeson, Christopher N .; Коуэн, Мортон Дж. (2011). «HLA-совместимая трансплантация костного мозга брату и сестре при большой β-талассемии» . Кровь
.
117
(5): 1745–1750. DOI : 10.1182 / кровь-2010-09-306829 . ISSN 0006-4971 . PMC 3056598 . PMID 21119108 . - «Генная терапия показывает перспективу лечения бета-талассемии и серповидноклеточной болезни» . 2012-03-28 . Проверено 15 октября 2015 .
- Уранюс, Селман. «Спленэктомия при гематологических нарушениях» . NCBI
. Проверено 4 апреля 2021 года . - А, Коэн. «Переливание крови при большой β-талассемии» . NCBI
. Проверено 4 апреля 2021 года . - CRISPR Therapeutics и Vertex Pharmaceuticals принимают меры для начала первого клинического испытания CRISPR / Cas9 в Европе в 2018 году. Клара Родригес Фернандес, 13 декабря 2021 г.
- Перейти
↑ Cappellini, Maria Domenica (2007). «Exjade® (деферасирокс, ICL670) в лечении хронической перегрузки железом, связанной с переливанием крови» .
Терапия и управление клиническими рисками
.
3
(2): 291–299. DOI : 10.2147 / tcrm.2007.3.2.291 . ISSN 1176-6336 . PMC 1936310 . PMID 18360637 . - Advani, Пуджа. «Лекарство от бета-талассемии» . Medscape
. Проверено 4 апреля 2021 года . - Шварц, М. Уильям (2012). Педиатрическая консультация за 5 минут . Липпинкотт Уильямс и Уилкинс. ISBN 9781451116564.
- Порвит, Анна; Маккалоу, Джеффри; Эрбер, Венди Н. (27 мая 2011 г.). Патология крови и костного мозга . Elsevier Health Sciences. ISBN 978-0702045356.
- Гемоглобинопатии . Издательство Jaypee Brothers. 2006. ISBN 9788180616693.
- Торре, Дарио М .; Лэмб, Джеффри С .; Руисвик, Джером Ван; Шапира, Ральф М. (2009). Кочара «Клиническая медицина для студентов» . Липпинкотт Уильямс и Уилкинс. ISBN 9780781766999.
- Бриссо, Пьер; Каппеллини, Мария Доменика (2014). «ЗАБОЛЕВАНИЕ ПЕЧЕНИ» . Международная федерация талассемии. Цитировать журнал требует |journal=( помощь )
- «ВОЗ | Глобальная эпидемиология нарушений гемоглобина и производные индикаторы обслуживания» . www.who.int
. Проверено 26 мая 2015 . - Берг, Шери; Биттнер, Эдвард А. (2013-10-16). Обзор медицины интенсивной терапии MGH . Липпинкотт Уильямс и Уилкинс. ISBN 9781451173680.
- Гематология стала проще . АвторДом. 2013-02-06. ISBN 9781477246511.
- Abouelmagd, Ахмед; Агели, Хусейн М. (2013). Основы генетики: праймер, охватывающий молекулярный состав генетического материала, экспрессию генов и генную инженерию, а также мутации и генетику человека . Универсальные издатели. ISBN 9781612331928.
- Weatherall, David J (2010). «Глава 47. Талассемии: нарушения синтеза глобина» . В Лихтмане, Массачусетс; Киппс, TJ; Seligsohn, U; Каушанский, К; Прчал, Дж. Т. (ред.). Талассемии: нарушения синтеза глобина
.
Гематология Уильямса
(8-е изд.). Компании McGraw-Hill. - «Статистика о талассемии» . Правильный диагноз
. Проверено 4 апреля 2021 года . - «Талассемия: генетическое заболевание крови, как ожидается, удвоится в следующие несколько десятилетий» . ScienceDaily
. Проверено 4 апреля 2021 года .
Лечение талассемии
Определяется типом и степенью тяжести. При умеренно выраженных симптомах лечение не назначают. Время от времени проводят только переливание крови. В основном это нужно после операций, родов или для предотвращения возможных осложнений. Люди с бета-талассемией требуют более частых переливаний крови. Для нормализации избыточного уровня железа им также назначают специфические препараты, которые связывают и выводят железо.
При выраженной и тяжелой формах болезни существует два способа лечения:
- частые переливания крови (раз в несколько недель), которые сочетают с приемом препаратов, выводящих лишнее железо из организма;
- пересадка костного мозга – единственный метод, который помогает полностью излечить человека от талассемии. К сожалению, далеко не всегда трансплантация бывает успешной.
Лекарственные препараты при талассемии назначают только для коррекции симптомов и осложнений. Медикаментозной терапии самого заболевания не существует.
Сколько стоит «Зинтегло»?
P. S. «Блюбёрд» выставила стоимость «Зинтегло» в размере 1,575 млн евро (1,77 млн долларов). Как и предполагалось, оплата будет осуществляться в рассрочку на протяжении пяти лет равными долями: вначале аванс, а затем ежегодные платежи, но только в том случае, если зафиксирована результативность лечения.
«Блюбёрд» еще не определилась с конечной стоимостью своей генотерапии, но она, очевидно, окажется запредельной. Понятное дело, в высокоразвитых странах со страховой медициной никто из пациентов не платит всю сумму из собственного кармана.
Да, по подсчетам «Блюбёрд», «справедливая цена» «Зинтегло», рассчитанная с учетом улучшения качества и продолжительности жизни пациента, находится в пределах 2,1 млн долларов, но, как ожидается, итоговая стоимость будет существенно меньше. Указанная астрономическая сумма исходит, очевидно, из тех совокупных затрат системы здравоохранения на многолетнюю хроническую терапию бета-талассемии. Так, по оценкам экспертов, один американский пациент с этим заболеванием обходится где-то в 200–300 тыс. долларов ежегодно.
Предложено в первый год внести 20% авансом, а оставшуюся сумму выплачивать 20-процентным частями в рассрочку ежегодно в течение четырех лет, причем при условии, что эффективность лечения удовлетворяет предопределенным критериям, таким как исчезновение необходимости в переливаниях крови или существенное снижение числа таких процедур.
Другими словами, платить придется лишь по факту результативности генотерапии, тем более пока нет неоспоримых данных о продолжительности сохранения ее целительного действия. Подход кажется разумным, если принимать во внимание различия в промежуточных исходах среди больных, хотя общая эффективность «Зинтегло» и правда впечатляет.
Согласно прогнозам отраслевых экспертов, стоимость «Зинтегло» составит 1,2 млн долларов в США и 900 тыс. долларов (785 тыс. евро) в Европе.
Осложнения талассемии
Возможные осложнения заболевания:
- излишек железа, которое входит в состав гемоглобина. Накопление этого элемента в организме приводит к поражению сердца, печени, эндокринной системы;
- подверженность инфекциям. Особенно актуально для пациентов, которым удалили селезенку;
- деформация костей, связанная с увеличением объема костного мозга. Чаще всего этот процесс затрагивает кости черепа, реже конечностей. Они истончаются и чаще ломаются;
- спленомегалия — увеличение селезенки, где в основном и гибнут дефектные эритроциты. Если селезенка увеличена очень сильно, ее удаляют. Эта операция называется спленэктомия;
- задержка роста и полового созревания;
- сердечные болезни (хроническая сердечная недостаточность и аритмии) могут развиться при тяжелом течении заболевания.
Дальнейшее чтение [ править ]
- Цао, Антонио; Галанелло, Ренцо (2010). «Бета-талассемия» . В Пагоне, Роберта А; Bird, Thomas D; Долан, Синтия Р.; Стивенс, Карен; Адам, Маргарет П. (ред.). GeneReviews . Вашингтонский университет, Сиэтл. PMID 20301599 .
- Бахал, Раман; Макнер, Николь Али; Кихано, Элиас; Лю, Яньфэн; Сулковски, Паркер; Турчик, Одри; Лу, И-Цзянь; Bhunia, Dinesh C .; Манна, Арунава; Greiner, Dale L .; Brehm, Michael A .; Ченг, Кристофер Дж .; Лопес-Хиральдес, Франсеск; Риккарди, Адель; Белоор, Джагадиш; Краузе, Дайан С .; Кумар, Прити; Галлахер, Патрик Дж .; Брэддок, Деметриос Т .; Зальцман, В. Марк; Ly, Danith H .; Глейзер, Питер М. (26 октября 2021 г.). «Коррекция in vivo анемии у β-талассемических мышей с помощью γPNA-опосредованного редактирования генов с доставкой наночастиц» . Nature Communications
.
7
: 13304. Bibcode : 2016NatCo … 713304B . DOI : 10.1038 / ncomms13304 . ISSN 2041-1723. PMC 5095181 . PMID 27782131 .
Гомозигота дает большую болезнь
Симптомы бета-талассемии определяются дисбалансом продукции
и
изменением количества бета-цепей протеиновой части гемоглобина
, что, главным образом, и предрасполагает к неэффективному кроветворению. Красные клетки крови, не вышедшие из ядерной стадии, массово находят смерть на месте своего рождения – в костном мозге, взрослые кровяные тельца (и в меньшей степени ретикулоциты) преждевременно заканчивают свою жизнь в селезенке, вследствие чего формируются симптомы выраженного малокровия. Кроме этого, в печени и селезенке образуются новые источники кроветворения. Кроветворение большой интенсивности, происходящее в костной системе, нарушает ее развитие (искажения и деформации), а состояние выраженной гипоксии влечет задержку общего развития детского организма. Такой вариант течения преимущественно присутствует у гомозиготной талассемии, нежели у гетерозиготной формы.
Симптомы, обусловленные участием двух родителей
Большая талассемия заявляет о себе уже от рождения маленького человека, правда, в первые месяцы выраженность симптомов не столь очевидна, однако ближе к годовалому возрасту, в крайнем случае – в начале второго года жизни, клинические проявления у детей приобретают довольно яркую окраску:
- Нельзя не заметить неестественную форму черепа (башенная, можно сказать, квадратная голова), больших размеров верхнюю челюсть и своеобразные черты лица ребенка: монголоидный тип, маленькие глазки, заметно приплющенная переносица (R-графия показывает патологические изменения);
- Притягивает внимание бледность кожных покровов, изменение их цвета – от сероватого до желтушного;
- У детей рано увеличивается печень и селезенка, что обусловлено экстрамедуллярным (внекостномозговым) типом кроветворения и неконтролируемым отложением в тканях гемосидерина (он образуется при распаде красных клеток крови);
- Дети часто болеют, поскольку снижается уровень иммунной защиты, они отстают в физическом развитии, у детей, которым повезло иметь более легкую форму гомозиготной бета-талассемии и суждено дожить до подросткового и взрослого состояния, задерживается развитие вторичных половых признаков;
- Обязательный в подобных случаях лабораторный контроль показывает соответствие клинических проявлений тяжелой гемолитической анемии.
Анализы
Ввиду того, что основным диагностическим критерием талассемии являются лабораторные анализы, то есть необходимость дать краткую характеристику картине крови, которая складывается при данной патологии:
- Уровень гемоглобина снижен до 50-30 г/л;
- Цветной показатель при расчетах показывает неутешительный результат – 0,5 и ниже;
- Эритроциты в мазке гипохромные, размеры клеток изменены (выраженный анизоцитоз), отмечается свойственная бета-талассемии (и мембранопатии) мишеневидность и базофильная зернистость;
- Осмотическая резистентность красных кровяных телец увеличена;
- Количество ретикулоцитов повышено;
- Биохимические показатели крови (билирубин за счет свободной фракции, сывороточное железо) повышены. Избыточные накопления железа способствуют формированию цирроза печени, приводят к развитию сахарного диабета и поражению сердечной мышцы.
Подтверждающим диагноз признаком в анализах крови пациента является увеличенное содержание фетального гемоглобина в красных клетках крови (до 20-90%).
Лечение
Выраженное кислородное голодание тканей и интенсивная работа кроветворной системы в костях (там, где у здорового человека ее нет) – показания к переливанию полноценных, способных обеспечить дыхание тканей, донорских эритроцитов. У больных большой талассемией гемотрансфузии с младенческих лет являются основным терапевтическим методом воздействия на болезнь. Вместе с тем, следует понимать, что переливание чужой крови не принадлежит к обычным лекарствам (таблеткам и растворам), поэтому подходить к такому лечению приходится с особой осторожностью. Тем не менее:
- На первом этапе больному обеспечивают ударный курс гемотрансфузий (в течение 2-3 недель больной получает до 10 переливаний, что позволяет повысить уровень гемоглобина до 120-140 г/л);
- На следующем этапе количество гемотрансфузий снижают, однако стараются держать Hb в пределах 90-100 г/л.
Подобная тактика лечения дает возможность:
- Заметно улучшить самочувствие;
- Снизить негативное воздействие патологии на костную систему;
- Повысить иммунитет;
- Препятствовать дальнейшему увеличению селезенки;
- Благоприятно влиять на физическое развитие малыша.
Однако не глядя на то, что переливание цельной крови полностью ушло в небытие (в таких случаях ее заменила эритроцитная масса), «неродная» биологическая среда все же сохраняет риск осложнений:
- Пирогенные реакции (они возникают преимущественно по причине нарушения техники отмывания красных клеток крови);
- Избыточное отложение гемосидерина (железосодержащего пигмента), которое способствует увеличению печени, развитию сердечной патологии и сахарного диабета.
Для того, чтобы обеспечить выведение из организма лишнего железа, больному назначают Десферал в виде внутримышечных инъекций (доза препарата соответствует возрасту и объему перелитой эрмассы). Довольно полезно в таких ситуациях сочетание Десферала с витамином С (аскорбиновой кислотой).
Значительное увеличение селезенки, а также сопутствующие изменения со стороны крови (лейкопения, тромбоцитопения) дают основание для удаления постоянно «растущего» органа (спленэктомии).
Между тем, стойкого долгосрочного улучшения состояния у больных гомозиготной β-талассемией можно добиться с помощью трансплантации костного мозга, однако операция эта представляет определенные трудности в плане подбора пары «донор-реципиент» и чревата развитием реакций после пересадки.
Напоследок – несколько слов о снижении продукции α-цепей
Продукция α-цепей красного кровяного пигмента находится под контролем двух пар генов, что делает возможным формирование разных форм заболевания:
- Если альфа-цепи отсутствуют полностью, то в эмбриональном периоде развития ребенка красный пигмент крови вообще не образовывается. В таких случаях исход один – водянка головного мозга и гибель ребенка;
- Если же нарушаются функциональные способности 1 или 2 генов из двух пар, то симптомы болезни начинают очень сильно напоминать клинические проявления β-талассемии, хотя показатели лабораторной диагностики в плане присутствия HbF и HbA2 – существенно разнятся, уровень этих гемоглобинов не имеет тенденции к повышению.
Терапия α-талассемии, в общем-то, ничем не отличается от лечебных мероприятий, используемых для воздействия на гетерозиготный вариант β-талассемии.
Уход
Лечение альфа-талассемии может включать переливание крови для поддержания гемоглобина на уровне, уменьшающем симптомы анемии. Решение о начале переливания зависит от клинической тяжести заболевания. [19] Спленэктомия — это возможный вариант лечения для повышения уровня общего гемоглобина в случаях обострения анемии из-за сверхактивной или увеличенной селезенки, или когда трансфузионная терапия невозможна. [20] Однако спленэктомии следует избегать, когда доступны другие варианты, из-за повышенного риска серьезных инфекций и тромбозов . [20]
Кроме того, камни в желчном пузыре могут быть проблемой, требующей хирургического вмешательства. Следует контролировать вторичные осложнения после приступа лихорадки , и большинство людей живут без какого-либо лечения. [1] [4]
Кроме того, трансплантацию стволовых клеток следует рассматривать как лечение (и лекарство), которое лучше всего проводить в раннем возрасте. Другие варианты, такие как генная терапия , все еще разрабатываются. [21]
Исследование Kreger et al, объединяющее ретроспективный обзор трех случаев большой альфа-талассемии и обзор литературы 17 случаев, показало, что внутриутробное переливание крови может привести к благоприятным результатам. В конечном итоге успешная трансплантация гемопоэтических клеток была проведена у четырех пациентов. [22]