Талассемия — это не одна, а группа похожих болезней, передающихся по наследству и вызванных нарушенным синтезом белковой части гемоглобина. Заболевание достается ребенку с вероятностью в 100%, если один из родителей содержит измененный ген. Поэтому оно считается самой распространенной наследственной патологией.
Наибольшее число случаев зарегистрировано в странах, расположенных по берегам Средиземного моря, на африканском континенте, в Средней Азии, Индии. Среди населения Азербайджана каждый десятый имеет измененные гены.
Немного истории
Болезнь была впервые описана в 1925 году американскими врачами-педиатрами, которые при лечении эмигрантов из Италии выявили одинаковую клиническую картину у детей с тяжелым малокровием, увеличением печени и селезенки, изменениями костей.
Затем появились работы, описывающие более легкое течение болезни у взрослых пациентов. Термин «талассемия» предложили в 1936 году. Дословно он означает «болезнь морского побережья». Высказана мысль о связи патогенеза заболевания с нарушением синтеза глобиновых цепочек.
Почему разрушаются эритроциты?
Патогенез талассемии достаточно изучен. Первичные изменения начинаются с нарушенного синтеза гемоглобина, а именно белковых цепочек вещества. Известно, что гемоглобин, входящий в состав 90% массы эритроцитов, — единственное вещество, способное связывать молекулы кислорода и разносить их из легочной ткани по всему организму.
Его структура состоит из пигмента (гема), включающего железо, и набора из двух пар белковых цепочек. Их называют по типичному расположению аминокислот альфа- и бета-цепями. При нарушенном синтезе одного из типов полипептидов накапливается другой вид.
В результате разрушаются все клетки эритроцитарного ряда (сами эритроциты и их предшественники), из них выходит гемоглобин. Малокровие (анемия) развивается по гипохромному типу. Это подтверждается низким цветовым показателем.
«Виновниками» разрушений являются гены, ответственные за построение белковой части гемоглобина. Их мутация нарушает способность составлять необходимый набор аминокислот в цепи. Причину изменений связывают с возбудителем малярии — плазмодием. Доказаны его мутирующие способности. Талассемия по территории распространенности совпадает с эпидемическими зонами малярии.
Основные четыре цепочки белков, связывающие гем и влияющие на эритроциты
Дети могут получить болезнь путем наследования от родителей в двух видах:
- гомозиготном — ген-мутант передается от обоих родителей;
- гетерозиготном — ген болезни передается только от матери или отца, носителем может быть один из родителей.
Соответственно, называются формы талассемии.
При гетерозиготном носительстве выделяют:
- «немой» ген (α-th2);
- «манифестный» ген (α-th1).
Наследование и эпидемиология
При этом в мире всего пять процентов населения – носители мутаций в генах цепей гемоглобина, а из них симптомам болезни подвержены чуть менее двух процентов людей. Стоит также учитывать, что жители разных регионов сталкиваются с талассемией с разной частотой. Прежде всего болезнь поражает жителей Средиземноморья, а также стран Центральной и Южной Азии. В России носителей мутаций, связанных с талассемией, сравнительно мало – около одного процента (для бета-типа).
Носители мутации могут даже не подозревать о своем статусе – часто повреждение лишь одной копии гена не ведёт к тяжелым симптомам. Однако носители мутации рискуют передать болезнь детям: если два носителя заведут ребенка, он может получить от родителей сразу обе копии мутировавшего гена с вероятностью около 25 процентов.
В предыдущем разделе мы уже обсуждали, что мутации в двух копиях генов приводит к более тяжелой форме талассемии. Но если посчитать вероятность такого события получится лишь около сотой процента.
Разновидности заболевания
Все виды заболевания связаны с кислородной недостаточностью, наступившей вследствие разрушения эритроцитов — единственных клеток, обеспечивающих газообмен в органах и тканях.
В зависимости от сбоя синтеза полипептидной цепочки различают:
- альфа-талассемию;
- бета-талассемию.
Наиболее распространена бета-талассемия. Она характеризуется избыточным накоплением α-цепочек глобина.
Выявлены также редкие формы болезни, которые названы гамма- и дельта-талассемией. В группу включены:
- некоторые гемоглобинопатии;
- гомозиготная альфа-талассемия с водянкой плода;
- смешанный вид β- и δ-талассемии, когда поражаются одновременно дельта- и бета-цепи.
Каждая форма имеет свои типичные клинические проявления и отличия. Но в зависимости от тяжести течения принято выделять:
- хроническую легкую — люди доживают до старости;
- хроническую среднетяжелую — пациенты не переживают детский возраст;
- тяжелую — ребенок погибает в период новорожденности.
Причины
Главная причина талассемии – наследственный фактор. При котором у одного пациента функционирует патологическая РНК (участвует в кодировании, чтении и регуляции генов), а у другого – ДНК (появляются изменения в хромосоме). На фоне этих изменений уменьшается или полностью прекращается синтез одной из цепочек Hb.
В норме количество альфа, бета-цепей – одинаково, поэтому изменение синтеза хотя бы одной из них приводит к дисбалансу и патологии.
Болезнь встречается как у девочек, так и у мальчиков.
Частота появлениябольшой талассемии 1:100 000, если оба родители носители мутационного гена.
Также на патогенез талассемии влияет и этническая принадлежность, так как чаще всего данная генетическая патология встречается у жителей Африки, Восточного Средиземноморья и Юго-Восточной, Центральной Азии. Определено это тем, что мутационный ген имеет большую сопротивляемость к малярии (обычно в африканских регионах) и большим количеством родственных браков, что приводит к разным генетическим нарушениям, включая талассемию.
В
Украине талассемия– редкая патология.
Всемирная организация здравоохранения приняла международную классификацию заболеваний 10-го пересмотра (мкб-10), как единый нормативный документ для ведения учета болезней, причин обращений пациентов в медицинские учреждения и причин смертности.
Согласно мкб талассемии (D56) имеет следующую классификацию:
- D56.0 – альфа-талассемия;
- D56.1 – бета-талассемия;
- D56.2 – дельта-бета талассемия;
- D56.3 – носитель талассемии;
- D56.9 – талассемия неуточненная.
Обновление кодов (мкб-11) будет вводиться в действие с 2022 года.
Клинические проявления большой талассемии
Симптомы талассемии зависят по клинике и времени проявления от вида генной мутации.
Большая талассемия (болезнь Кули) развивается при гомозиготной передаче от родителей. Ее проявления заметны у детей сразу после рождения.
У новорожденного замечают:
- удлиненный вверх череп («башенный»);
- более развитую верхнюю челюсть;
- монголоидный тип лицевого скелета.
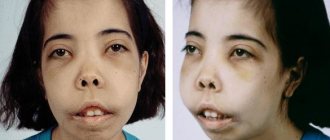
Типичная внешность и изменения структуры костей черепа используются в диагностике
К 12-месячному возрасту проявляется:
- расширенная носовая перегородка, «приплюснутый» нос;
- костные наросты на стопах;
- нарушенный прикус;
- желтушность кожи (в связи с поражением селезенки).
Кислородное голодание тканей приводит к органным нарушениям. При осмотре отмечаются:
- увеличение печени, край плотный на ощупь (развивается ранний цирроз);
- возникновение сердечной недостаточности (лишнее железо откладывается в миокарде и поражает сократимость сердечной мышцы);
- отставание в физическом и умственном развитии от сверстников.
У малыша развивается сахарный диабет из-за фиброзирования поджелудочной железы и печеночная недостаточность. В тяжелых случаях он живет не более одного года.
В старшем возрасте проявляются такие осложнения:
- трофические язвы на коже, вызванные локальными нарушениями кровообращения;
- частые переломы костей;
- повторные воспаления в легких;
- калькулезный холецистит;
- кардиосклероз;
- сепсис при контакте с инфекцией;
- нарушенное полового развитие.
Выделяют промежуточную форму β-талассемии с более доброкачественным течением. Она начинается у детей старшего возраста. Внешний вид ребенка не изменяется. Нет отставания в умственном и физическом развитии. Основные проявления вызываются увеличением селезенки и повышенной ломкостью костной ткани.
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников — бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Клиника малой талассемии
Малая или гетерозиготная форма передается одним из родителей. Второй «здоровый» ген сглаживает повреждения. Симптомы талассемии могут длительно не проявляться или вызывать признаки, общие с другими заболеваниями:
- слабость, повышенную утомляемость;
- головные боли;
- головокружение.
При осмотре обращается внимание на следующее:
- бледность кожи с желтушным оттенком;
- возможно увеличение печени и селезенки.
У ребенка с малой формой резко увеличена восприимчивость к инфекциям, снижен иммунитет. Очень тяжело и часто протекают вирусные и кишечные инфекционные заболевания.
Во взрослом состоянии у женщины при беременности развивается водянка плода (повышение внутричерепного давления и скопление жидкости в желудочках мозга). Эта патология несовместима с жизнью ребенка, в случае нормальных родов ведет к тяжелым неврологическим и психическим отклонениям.
Минимальной β-талассемией называют синдром Сильвестрони — Бьянко. Эта форма заболевания протекает практически бессимптомно, обнаруживается случайно в семьях со случаями талассемии.
Диагностика
Диагностика талассемии должна начинаться с генетической консультации супругов перед зачатием ребенка. Во время беременности при необходимости проводят анализ амниотической жидкости. Уже на ранних сроках в эритроцитах плода можно обнаружить характерные изменения.
Порой внешний осмотр и выяснение семейного анамнеза позволяют заподозрить болезнь у ребенка без лабораторной диагностики.
В анализе крови выявляют:
- снижение уровня гемоглобина до 30–50 г/л при гомозиготном варианте и до 90 –110 г/л при гетерозиготном;
- низкий цветовой показатель (менее 0,5), который образуется малым насыщением клеток гемоглобином);
- рост ретикулоцитов (предшественников эритроцитов) до 4%.
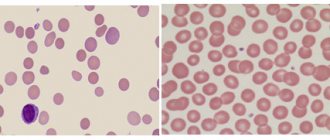
При просмотре окрашенного мазка под микроскопом обращают внимание на следующее:
- наличие слабо окрашенных (гипохромных) эритроцитов;
- изменение размеров и формы красных клеток, эритроциты приобретают формы овала, серпа, шара.
Одна из характерных разновидностей серповидных клеток — дрепаноциты.
Дрепаноциты образуются при любых гемоглобинопатиях, содержат особую разновидность (S гемоглобин), который может в условиях низкой концентрации образовывать полимеры и изменять форму оболочки клетки
Биохимические тесты показывают нарушенный обмен железа, как при гемолитической анемии:
- высокий уровень свободного билирубина;
- повышенную концентрацию железа в сыворотке;
- сниженную способность к связыванию железа.
Исследование особенностей гемоглобина проводят с помощью ацетат-целлюлозной пленки. Подобный анализ позволяет определить количественный уровень фракций. Гомозиготная бета-талассемия отличается увеличенным уровнем фетального гемоглобина (в норме он содержится у плода, а у взрослого человека всего до 1%), неспособного переносить кислород.
Использование метода полимеразной цепной реакции позволяет изучить строение полипептидных цепочек гемоглобина.
Генетический анализ выявляет мутацию в одиннадцатой паре хромосом при β-талассемии, другие специфические изменения, типичные для прочих форм.
Исследование пунктата костного мозга проводится для выявления повышенного содержания незрелых эритроцитов в виде сидеробластов.
Рентгенологическое исследование способствует обнаружению дефектов костной ткани:
- участков остеопороза (сниженной плотности);
- увеличенной массы костей черепа;
- поперечной исчерченности на мелких костях кистей и стоп;
- на рентгенограмме черепа при большой форме β-талассемии видно типичное игольчатое поражение надкостницы, которое называется симптомом «волосатого черепа».
Ультразвуковое исследование подтверждает увеличение печени и селезенки, помогает обнаружить камни в желчевыводящей системе.
Лабораторные данные более отчетливо выражены при β-талассемии. Другие формы болезни не дают четкой картины.
Заболевания, с которыми необходимо проводить дифференциальную диагностику:
- железодефицитная анемия,
- серповидно-клеточная анемия,
- гемолитическая аутоиммунная анемия,
- наследственный микросфероцитоз.
использованная литература
- ^ a b c d e f g h Орига, Рафаэлла; Мои, Паоло; Галанелло, Ренцо; Цао, Антонио (1 января 1993 г.). «Альфа-талассемия» . GeneReviews
. PMID 20301608 . Проверено 22 сентября 2021 года .обновление 2013 - ^ a b Патология BRS (4-е изд.). Lippincott Williams & Wilkins medical. Декабрь 2009 г. с. 162. ISBN. 978-1451115871.
- ^ a b c d e «Обследование альфа-талассемии: подходы, лабораторные исследования, электрофорез гемоглобина» . emedicine.medscape.com
. Дата обращения 24 мая 2021 . - ^ a b «Осложнения и лечение | Талассемия | Заболевания крови | NCBDDD | CDC» . www.cdc.gov
. Проверено 22 сентября 2021 года . - Интернет-Менделирующее наследование в человеке (OMIM): гемоглобин — альфа-локус 1; HBA1 — 141800
- Интернет-Менделирующее наследование в человеке (OMIM): гемоглобин — альфа-локус 2; HBA2 — 141850
- Руководство Ланцковского по детской гематологии и онкологии 6-е издание (2016) .
- ^ a b c d e «Альфа-талассемия — Симптомы, диагностика и лечение | BMJ Best Practice» . bestpractice.bmj.com
. Дата обращения 17 ноября 2021 . - Ссылка, Genetics Home. «Альфа-талассемия» . Домашний справочник по генетике
. Проверено 25 ноября 2021 года . - Орига, Рафаэлла; Мои, Паоло (1993), Адам, Маргарет П .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Альфа-талассемия» , GeneReviews®
, Вашингтонский университет, Сиэтл, PMID 20301608 , получено 25 ноября 2021 г. - «Оценка анемии — Этиология | BMJ Best Practice» . bestpractice.bmj.com
. Проверено 25 ноября 2021 года . - Steensma DP, Gibbons RJ, Хиггс DR (январь 2005). «Приобретенная альфа-талассемия в сочетании с миелодиспластическим синдромом и другими гематологическими злокачественными новообразованиями» . Кровь
.
105
(2): 443–52. DOI : 10.1182 / кровь-2004-07-2792 . PMID 15358626 . - ^ Б Galanello R, Cao A (февраль 2011). «Обзор генного теста. Альфа-талассемия» . Генетика в медицине
.
13
(2): 83–8. DOI : 10.1097 / GIM.0b013e3181fcb468 . PMID 21381239 . - «Болезнь гемоглобина H» . Сиротка
. Проверено 22 сентября 2021 года . - Vichinsky EP (1 января 2009). «Большая альфа-талассемия — новые мутации, внутриутробное ведение и исходы». Гематология.Американское общество гематологии.Образовательная программа
.
2009
(1): 35–41. DOI : 10,1182 / asheducation-2009.1.35 . PMID 20008180 . - Songdej D, Babbs C, Хиггс DR (март 2017). «Международный регистр выживших с синдромом фетальной водянки Барта» . Кровь
.
129
(10): 1251–1259. DOI : 10,1182 / кровь 2016-08-697110 . PMC 5345731 . PMID 28057638 . - Кеохан, E; Смит, L; Валенга, Дж (2015). Гематология Родака: клинические принципы и применение (5-е изд.). Elsevier Health Sciences. п. 466. ISBN. 978-0-323-23906-6.
- ^ а б в Випракасит, VIP; Экваттанакит, Супачай (1 апреля 2018 г.). «Клиническая классификация, скрининг и диагностика талассемии». Гематологические / онкологические клиники Северной Америки
. Талассемия.
32
(2): 193–211. DOI : 10.1016 / j.hoc.2017.11.006 . ISSN 0889-8588 . PMID 29458726 . - «UpToDate» . www.uptodate.com
. Проверено 25 ноября 2021 года . - ^ а б Тахер, Али; Мусаллам, Халед; Каппеллини, Мария Доменика, ред. (2017). Руководство по ведению нетрансфузионно-зависимой талассемии (NTDT) (2-е изд.). Международный фонд талассемии. С. 24–32 . Дата обращения 5 ноября 2021 .
- «Талассемия | Врач | Пациент» . Пациент
. Проверено 22 сентября 2021 года . - Крегер Э.М., Зингер С.Т., Витт Р.Г., Свитерс Н., Лианоглу Б., Лал А. и др. (Декабрь 2021 г.). «Благоприятные исходы после внутриутробного переливания у плодов с большой альфа-талассемией: серия случаев и обзор литературы». Пренатальная диагностика
.
36
(13): 1242–1249. DOI : 10.1002 / pd.4966 . PMID 27862048 . S2CID 29734120 . - Harteveld CL, Хиггс DR (май 2010). «Альфа-талассемия» . Журнал «Орфанет редких болезней»
.
5
(1): 13. DOI : 10,1186 / 1750-1172-5-13 . PMC 2887799 . PMID 20507641 . - Гематология стала проще. АвторДом. 2013-02-06. ISBN 9781477246511. Стр. 246
Генетическая интерпретация видов бета-талассемии
Ученые-генетики выяснили интересную закономерность: люди имеют одинаковую мутацию генов, отвечающих за синтез гемоглобина, но клиника и степень выраженности заболевания у них отличаются.
Гены могут находиться в следующих состояниях:
- нормальные — характерны для здорового человека;
- поврежденные частично — «работает» неполно, из-за чего синтез полипептидных цепей недостаточен;
- разрушенные полностью — синтез останавливается.
По этому признаку виды талассемии делят на:
- минор — самая легкая форма, поврежден только один ген, внешне человек выглядит здоровым, по анализу крови можно предположить небольшую анемию;
- интермедиа — недостаток бета-цепей серьезно сказывается на синтезе, эритроциты недоразвиты, малокровие выражено с явными признаками, но организм еще может приспособиться, поэтому отсутствует необходимость в постоянных переливаниях крови;
- майор — мутации подверглись все гены, больному необходимы постоянные переливания крови по жизненным показаниям.
Как работает «Зинтегло»?
Лечение при помощи генно-терапевтического «Зинтегло», ранее называвшегося «Лентиглобин» (LentiGlobin), предполагает однократную инъекцию, после которой пациенты с бета-талассемией получают очень высокий шанс навсегда избавиться от необходимости в переливаниях эритроцитарной массы.
Извлеченные аферезом гемопоэтические стволовые клетки CD34+ пациента трансдуцируются ex vivo путем лентивирусного переноса функциональных копий модифицированного гена бета-глобина (βA-T87Q-глобин). Затем клетки возвращаются в организм пациента, предварительно прошедшего химиотерапевтическую процедуру миелоаблативного кондиционирования костного мозга бусульфаном. После приживления стволовых клеток и их дифференцирования запускается синтез эритроцитов, содержащих биологически активный βA-T87Q-глобин, который сочетается с альфа-глобином, чтобы продуцировать функциональный гемоглоблин A. Как только последний выходит на нормальный уровень, можно рассчитывать, что исцеляющий эффект «Зинтегло» сохранится на протяжении всей жизни.
Генетические разновидности альфа-талассемии
По охвату мутацией генов, их локусов (определенных частей, отрезков), отвечающих за синтез альфа-цепочек полипептидов, предлагается выделение нескольких групп заболевания:
- мутация только в одном локусе — клинических проявлений нет;
- изменения двух (пары) локусов в одном или разных генах — анализ крови показывает снижение гемоглобина, уменьшение размера эритроцитов;
- поражение трех локусов — выражается в кислородной гипоксии органов, увеличении селезенки, возникновении Н гемоглобинопатии, образующийся гемоглобин нестоек, разлагаясь, вызывает гемолитическую анемию;
- мутация всех локусов — полностью прекращает синтез альфа-цепей, при подобной ситуации происходит внутриутробная гибель плода или ребенок умирает сразу после родов.
Генетические исследования подтвердили также неодинаковое значение пар генов, одна пара из двух является главной, а другая имеет второстепенную роль. Клинические проявления зависят от того, в какой паре произошла мутация.
Пересадка костного мозга считается наиболее результативным способом
Проблемы терапии
Лечение талассемии зависит от тяжести поражения эритропоэза, степени охвата генов процессами мутации. В настоящее время применяются следующие методы.
Диетический рацион направлен на снижение всасывания железа в кишечнике, рекомендуются орехи, какао, соя, чай.
Тяжелая форма требует регулярного переливания крови, эритроцитарной массы, размороженных и профильтрованных эритроцитов. Эффективность временная, возможны побочные эффекты, но главное — сохранить жизнь больному.
Терапия дополняется ежедневным устранением излишков железа с помощью введения (хелатов — специальных комплексов, повышающих действие лекарства). Для связывания железа назначается Десферал. Этот препарат предупреждает сидероз (патологическое состояние, вызванное отложением в тканях железа), но на уровень гемоглобина не влияет.
При возникновении резкого ухудшения состояния по аналогии с гемолитическими кризами показаны глюкокортикоиды в больших дозах.
Спленэктомия возможна при больших размерах селезенки детям после пятилетнего возраста. Наиболее оптимален возраст 8–10 лет. После удаления селезенки наступает период улучшения, но опасен риск присоединения инфекции.
Для трансплантации необходим донор, совпадающий по всем параметрам, лучше всего из близких родственников.
К симптоматическим средствам относятся препараты гепатопротекторного действия, большие дозы аскорбинки помогают вывести излишки железа из организма.
Все формы талассемии нуждаются в препаратах с фолиевой кислотой и витаминами группы В. На фоне присоединившейся инфекции, при беременности следует применять фолиевую кислоту в больших дозах, поскольку неэффективное кроветворения при талассемии значительно увеличивает ее потребление клетками.