Thalassemia is not one, but a group of similar diseases, inherited and caused by impaired synthesis of the protein part of hemoglobin. The disease is transmitted to a child with a 100% probability if one of the parents contains the altered gene. Therefore, it is considered the most common hereditary pathology.
The largest number of cases have been registered in countries located along the shores of the Mediterranean Sea, on the African continent, in Central Asia, and India. Among the population of Azerbaijan, every tenth person has altered genes.
A little history
The disease was first described in 1925 by American pediatricians, who, when treating emigrants from Italy, identified the same clinical picture in children with severe anemia, enlarged liver and spleen, and bone changes.
Then there were works describing a milder course of the disease in adult patients. The term thalassemia was coined in 1936. Literally it means “sea coast disease.” The idea has been expressed about the connection between the pathogenesis of the disease and a violation of the synthesis of globin chains.
Why are red blood cells destroyed?
The pathogenesis of thalassemia has been sufficiently studied. Primary changes begin with impaired synthesis of hemoglobin, namely the protein chains of the substance. It is known that hemoglobin, which makes up 90% of the mass of red blood cells, is the only substance capable of binding oxygen molecules and carrying them from the lung tissue throughout the body.
Its structure consists of a pigment (heme), which includes iron, and a set of two pairs of protein chains. They are called alpha and beta chains based on their typical arrangement of amino acids. When the synthesis of one type of polypeptide is impaired, another type accumulates.
As a result, all cells of the erythrocyte series (the erythrocytes themselves and their precursors) are destroyed, and hemoglobin is released from them. Anemia (anemia) develops according to the hypochromic type. This is confirmed by the low color index.
The “culprits” of the destruction are the genes responsible for the construction of the protein part of hemoglobin. Their mutation disrupts the ability to compose the required set of amino acids in the chain. The cause of the changes is associated with the causative agent of malaria - Plasmodium. Its mutating abilities have been proven. Thalassemia's prevalence coincides with the epidemic zones of malaria.
The main four protein chains that bind heme and influence red blood cells
Children can get the disease by inheriting from their parents in two forms:
- homozygous - the mutant gene is transmitted from both parents;
- heterozygous - the disease gene is transmitted only from the mother or father; one of the parents can be the carrier.
Accordingly, forms of thalassemia are called.
In heterozygous carriage there are:
- “silent” gene (α-th2);
- “manifest” gene (α-th1).
Inheritance and epidemiology
At the same time, only five percent of the world's population are carriers of mutations in the genes of hemoglobin chains, and of these, slightly less than two percent of people are susceptible to symptoms of the disease. It is also worth considering that residents of different regions experience thalassemia with different frequencies. The disease primarily affects residents of the Mediterranean region, as well as the countries of Central and South Asia. In Russia, there are relatively few carriers of mutations associated with thalassemia - about one percent (for the beta type).
Carriers of the mutation may not even be aware of their status - often damage to just one copy of the gene does not lead to severe symptoms. However, mutation carriers run the risk of passing the disease on to their children: if two carriers have a child, he can receive both copies of the mutated gene from his parents at once with a probability of about 25 percent.
In the previous section, we already discussed that mutations in two copies of genes lead to a more severe form of thalassemia. But if you calculate the probability of such an event, it turns out to be only about a hundredth of a percent.
Types of disease
All types of diseases are associated with oxygen deficiency, which occurs as a result of the destruction of red blood cells - the only cells that ensure gas exchange in organs and tissues.
Depending on the failure of the synthesis of the polypeptide chain, there are:
- alpha thalassemia;
- beta thalassemia.
The most common is beta thalassemia. It is characterized by excessive accumulation of globin α-chains.
Rare forms of the disease have also been identified, which are called gamma and delta thalassemia. The group includes:
- some hemoglobinopathies;
- homozygous alpha thalassemia with hydrops fetalis;
- mixed type of β- and δ-thalassemia, when delta and beta chains are affected simultaneously.
Each form has its own typical clinical manifestations and differences. But depending on the severity of the current, it is customary to distinguish:
- chronic lung - people live to old age;
- chronic moderate - patients do not survive childhood;
- severe - the child dies during the neonatal period.
Causes
The main cause of thalassemia is a hereditary factor. In which one patient has abnormal RNA (participates in coding, reading and regulation of genes), and another has abnormal DNA (changes appear in the chromosome). Against the background of these changes, the synthesis of one of the Hb chains decreases or completely stops.
Normally, the number of alpha and beta chains is the same, so a change in the synthesis of at least one of them leads to an imbalance and pathology.
The disease occurs in both girls and boys.
The incidence of thalassemia major is 1:100,000 if both parents are carriers of the mutation gene.
Ethnicity also influences the pathogenesis of thalassemia, since most often this genetic pathology occurs in residents of Africa, the Eastern Mediterranean and Southeast and Central Asia. This is determined by the fact that the mutation gene has greater resistance to malaria (usually in African regions) and a large number of consanguineous marriages, which leads to various genetic disorders, including thalassemia.
In
Ukraine, thalassemia is a rare pathology.
The World Health Organization has adopted the International Classification of Diseases, 10th revision (ICD-10), as a single normative document for keeping records of diseases, reasons for patient visits to medical institutions and causes of mortality.
According to the ICD thalassemia (D56) has the following classification:
- D56.0 – alpha thalassemia;
- D56.1 – beta thalassemia;
- D56.2 – delta-beta thalassemia;
- D56.3 – carrier of thalassemia;
- D56.9 – thalassemia, unspecified.
The code update (ICD-11) will be introduced in 2022.
Clinical manifestations of thalassemia major
The symptoms of thalassemia depend on the clinical picture and time of manifestation on the type of gene mutation.
Thalassemia major (Cooley's disease) develops through homozygous transmission from parents. Its manifestations are noticeable in children immediately after birth.
The newborn notices:
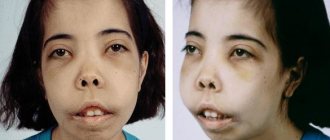
- skull elongated upward (“tower”);
- more developed upper jaw;
- Mongoloid type of facial skeleton.
Typical appearance and changes in the structure of the skull bones are used in diagnosis
By 12 months of age the following appears:
- widened nasal septum, “flattened” nose;
- bone growths on the feet;
- broken bite;
- yellowness of the skin (due to damage to the spleen).
Oxygen starvation of tissues leads to organ disorders. Upon examination the following are noted:
- enlarged liver, the edge is dense to the touch (early cirrhosis develops);
- the occurrence of heart failure (excess iron is deposited in the myocardium and affects the contractility of the heart muscle);
- lagging behind peers in physical and mental development.
The baby develops diabetes mellitus due to fibrosis of the pancreas and liver failure. In severe cases, it lives no more than one year.
At older ages, the following complications appear:
- trophic ulcers on the skin caused by local circulatory disorders;
- frequent bone fractures;
- repeated inflammation in the lungs;
- calculous cholecystitis;
- cardiosclerosis;
- sepsis upon contact with infection;
- impaired sexual development.
There is an intermediate form of β-thalassemia with a more benign course. It begins in older children. The child's appearance does not change. There is no lag in mental and physical development. The main manifestations are caused by an enlarged spleen and increased fragility of bone tissue.
Symptoms and signs of thalassemia
In most cases, thalassemia is detected at the stage of prenatal diagnosis. If necessary, treatment begins immediately, without waiting for symptoms to appear. If the disease is not detected by prenatal diagnosis, the following symptoms are expected:
- pallor or yellowness of the mucous membranes;
- slow growth;
- dark urine;
- abdominal enlargement;
- deformation of bones, especially the bones of the skull.
The time at which the first signs of thalassemia appear largely depends on the type of disease and the number of mutations. In some children, symptoms are registered soon after birth, in others - in the first two years of life.
Diagnosis of thalassemia
Symptoms of thalassemia are more or less characteristic. To make a final diagnosis, the doctor needs laboratory results. If thalassemia is suspected, a general blood test is required. It will show a reduced number of red blood cells that are small, light, different in shape and size. In addition to mature cells, the smear will contain many of their precursors - blasts. Additionally, other specific blood tests may be prescribed to determine the severity of the disorders (biochemical analysis, determination of iron-binding capacity of plasma or serum ferritin). Molecular tests (PCR) have also been developed to determine the presence of mutations.
Ultrasound is used to assess the condition of the liver and spleen, and radiography is used to identify bone tissue pathology.
In a child, thalassemia can be diagnosed during pregnancy. This study is especially recommended for parents who are sick or may be carriers of this disease. There are two diagnostic methods:
- chorionic villus biopsy - performed at the 11th week of pregnancy;
- amniocentesis (sampling of amniotic fluid) - prescribed at the 16th week.
Thalassemia Minor Clinic
The minor or heterozygous form is transmitted by one of the parents. The second “healthy” gene smoothes out damage. Symptoms of thalassemia may not appear for a long time or may cause symptoms common to other diseases:
- weakness, increased fatigue;
- headache;
- dizziness.
During inspection, pay attention to the following:
- pale skin with a jaundiced tint;
- possible enlargement of the liver and spleen.
A child with a minor form has a sharply increased susceptibility to infections and reduced immunity. Viral and intestinal infectious diseases are very severe and frequent.
In adulthood, a woman develops hydrops fetalis during pregnancy (increased intracranial pressure and accumulation of fluid in the ventricles of the brain). This pathology is incompatible with the life of the child; in the case of normal birth, it leads to severe neurological and mental disorders.
β-thalassemia minimalis is called Silvestroni-Bianco syndrome. This form of the disease is practically asymptomatic and is discovered incidentally in families with cases of thalassemia.
Diagnostics
Diagnosis of thalassemia should begin with genetic counseling of spouses before conceiving a child. During pregnancy, if necessary, an analysis of the amniotic fluid is performed. Already in the early stages, characteristic changes can be detected in the red blood cells of the fetus.
Sometimes an external examination and clarification of the family history make it possible to suspect a disease in a child without laboratory diagnosis.
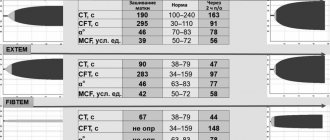
A blood test reveals:
- decrease in hemoglobin level to 30–50 g/l with the homozygous variant and to 90–110 g/l with the heterozygous variant;
- low color index (less than 0.5), which is formed by low saturation of cells with hemoglobin);
- growth of reticulocytes (precursors of red blood cells) up to 4%.
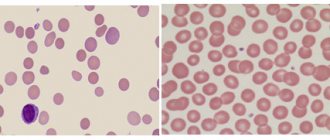
When viewing a stained smear under a microscope, pay attention to the following:
- the presence of weakly stained (hypochromic) red blood cells;
- changes in the size and shape of red cells, red blood cells take on the shape of an oval, sickle, or ball.
One of the characteristic types of sickle cells is drepanocytes.
Drepanocytes are formed in any hemoglobinopathies; they contain a special type (S hemoglobin), which can form polymers in conditions of low concentration and change the shape of the cell membrane
Biochemical tests show impaired iron metabolism, as in hemolytic anemia:
- high level of free bilirubin;
- increased serum iron concentration;
- reduced ability to bind iron.
The study of the characteristics of hemoglobin is carried out using cellulose acetate film. Such an analysis makes it possible to determine the quantitative level of fractions. Homozygous beta thalassemia is characterized by an increased level of fetal hemoglobin (normally it is found in the fetus, but in an adult it is only up to 1%), which is unable to carry oxygen.
The use of the polymerase chain reaction method makes it possible to study the structure of hemoglobin polypeptide chains.
Genetic analysis reveals a mutation in the eleventh pair of chromosomes in β-thalassemia and other specific changes typical of other forms.
A bone marrow punctate examination is carried out to identify an increased content of immature red blood cells in the form of sideroblasts.
X-ray examination helps to detect bone tissue defects:
- areas of osteoporosis (reduced density);
- increased mass of skull bones;
- transverse striations on the small bones of the hands and feet;
- X-rays of the skull in β-thalassemia major show a typical needle-like lesion on the periosteum, which is called the “hairy skull” sign.
An ultrasound examination confirms an enlarged liver and spleen and helps detect stones in the biliary system.
Laboratory findings are more pronounced in β-thalassemia. Other forms of the disease do not provide a clear picture.
Diseases that require differential diagnosis:
- Iron-deficiency anemia,
- sickle cell anemia,
- hemolytic autoimmune anemia,
- hereditary microspherocytosis.
References
- ^ abcdefgh Origa, Rafaella; Mine, Paolo; Galanello, Renzo; Cao, Antonio (1 January 1993). "Alpha thalassemia". GeneReviews
. PMID 20301608. Retrieved September 22, 2021.updated 2013 - ^ ab BRS Pathology (4th ed.). Lippincott Williams & Wilkins medical. December 2009 p. 162. ISBN. 978-1451115871.
- ^ abcde "Evaluation for alpha thalassemia: approaches, laboratory tests, hemoglobin electrophoresis". emedicine.medscape.com
. Retrieved May 24, 2021. - ^ ab “Complications and treatment | Thalassemia | Blood diseases | NCBDDD | CDC". www.cdc.gov
. Retrieved September 22, 2021. - Online Mendelian Inheritance in Man (OMIM): hemoglobin - alpha locus 1; HBA1 - 141800
- Online Mendelian Inheritance in Man (OMIM): hemoglobin - alpha locus 2; HBA2 - 141850
- Lanzkovsky's Guide to Pediatric Hematology and Oncology 6th edition (2016).
- ^ abcde "Alpha thalassemia - Symptoms, diagnosis and treatment | BMJ Best Practice". bestpractice.bmj.com
. Retrieved November 17, 2021. - Link, Genetics Home. "Alpha thalassemia". A Home Guide to Genetics
. Retrieved November 25, 2021. - Origa, Rafaella; Moi, Paolo (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (ed.), "Alpha Thalassemia", GeneReviews®
, University of Washington, Seattle, PMID 20301608, retrieved November 25, 2021. - "Assessment of Anemia - Etiology | BMJ Best Practice". bestpractice.bmj.com
. Retrieved November 25, 2021. - Steensma DP, Gibbons RJ, Higgs DR (January 2005). "Acquired alpha thalassemia in association with myelodysplastic syndrome and other hematologic malignancies". Blood
.
105
(2):443–52. DOI: 10.1182/blood-2004-07-2792. PMID 15358626. - ^B Galanello R, Cao A (February 2011). “Gene test review. Alpha thalassemia". Genetics in Medicine
.
13
(2): 83–8. DOI: 10.1097/GIM.0b013e3181fcb468. PMID 21381239. - "Hemoglobin H disease". Orphan
. Retrieved September 22, 2021. - Vichinsky EP (January 1, 2009). "Alpha thalassemia major—new mutations, in utero management and outcomes." Hematology. American Society of Hematology. Educational program
.
2009
(1): 35–41. DOI: 10.1182/asheducation-2009.1.35. PMID 20008180. - Songdej D, Babbs C, Higgs DR (March 2017). "International Registry of Survivors of Barth's Hydrops Fetal Syndrome". Blood
.
129
(10):1251–1259. DOI: 10.1182/blood 2016-08-697110. PMC 5345731. PMID 28057638. - Keohane, E; Smith, L; Walenga, J (2015). Rodak's Hematology: Clinical Principles and Applications (5th ed.). Elsevier Health Sciences. item 466. ISBN. 978-0-323-23906-6.
- ^ a b c Viprakasit, VIP; Equattanakit, Supachai (April 1, 2018). "Clinical classification, screening and diagnosis of thalassemia." Hematology/Oncology Clinics of North America
.
Thalassemia. 32
(2): 193–211. DOI: 10.1016/j.hoc.2017.11.006. ISSN 0889-8588. PMID 29458726. - "UpToDate" . www.uptodate.com
. Retrieved November 25, 2021. - ^ a b Taher, Ali; Musallam, Khaled; Cappellini, Maria Domenica, ed. (2017). Guidelines for the management of non-transfusion-dependent thalassemia (NTDT) (2nd ed.). International Thalassemia Foundation. pp. 24–32. Retrieved November 5, 2021.
- "Thalassemia | Doctor | Patient" . Patient
. Retrieved September 22, 2021. - Kroeger EM, Singer ST, Witt RG, Sweeters N, Lianoglou B, Lal A, et al (December 2021). "Favorable outcomes after intrauterine transfusion in fetuses with alpha thalassemia major: a case series and review of the literature." Prenatal diagnosis
.
36
(13):1242–1249. DOI: 10.1002/pd.4966. PMID 27862048. S2CID 29734120. - Harteveld CL, Higgs DR (May 2010). "Alpha thalassemia". Journal "Orphanet of Rare Diseases"
.
5
(1): 13. DOI: 10.1186/1750-1172-5-13. PMC 2887799. PMID 20507641. - Hematology made simpler. AuthorHouse. 2013-02-06. ISBN 9781477246511. Pg. 246
Genetic interpretation of beta thalassemia species
Genetic scientists have discovered an interesting pattern: people have the same mutation of the genes responsible for the synthesis of hemoglobin, but the clinical picture and severity of the disease differ.
Genes can be in the following states:
- normal - characteristic of a healthy person;
- partially damaged - “works” incompletely, which is why the synthesis of polypeptide chains is insufficient;
- completely destroyed - the synthesis stops.
On this basis, types of thalassemia are divided into:
- minor - the mildest form, only one gene is damaged, the person looks healthy outwardly, a blood test suggests slight anemia;
- intermedia - the lack of beta chains seriously affects the synthesis, red blood cells are underdeveloped, anemia is expressed with obvious signs, but the body can still adapt, so there is no need for constant blood transfusions;
- major - all genes have undergone mutations, the patient requires constant blood transfusions for health reasons.
How does Zinteglo work?
Treatment with the gene therapy Zynteglo, formerly called LentiGlobin, involves a single injection, after which patients with beta thalassemia have a very high chance of permanently eliminating the need for red blood cell transfusions.
Apheresis-extracted patient CD34+ hematopoietic stem cells are transduced ex vivo by lentiviral transfer of functional copies of the modified beta-globin gene (βA-T87Q-globin). The cells are then returned to the patient's body after undergoing a chemotherapy procedure called myeloablative bone marrow conditioning with busulfan. After the engraftment of stem cells and their differentiation, the synthesis of red blood cells containing biologically active βA-T87Q-globin is triggered, which combines with alpha globin to produce functional hemoglobin A. As soon as the latter reaches normal levels, the healing effect of Zynteglo can be expected will remain throughout life.
Genetic variants of alpha thalassemia
Based on the coverage of mutations in genes and their loci (certain parts, segments) responsible for the synthesis of alpha chains of polypeptides, it is proposed to distinguish several groups of the disease:
- mutation in only one locus - no clinical manifestations;
- changes in two (pairs) of loci in one or different genes - a blood test shows a decrease in hemoglobin, a decrease in the size of red blood cells;
- damage to three loci - is expressed in oxygen hypoxia of organs, enlargement of the spleen, the occurrence of H hemoglobinopathy, the resulting hemoglobin is unstable, decomposing, causing hemolytic anemia;
- mutation of all loci - completely stops the synthesis of alpha chains; in such a situation, intrauterine fetal death occurs or the child dies immediately after birth.
Genetic studies have also confirmed the unequal significance of pairs of genes, one pair of two is the main one, and the other has a secondary role. Clinical manifestations depend on which pair the mutation occurred in.
Bone marrow transplant is considered the most effective method
Therapy problems
Treatment of thalassemia depends on the severity of damage to erythropoiesis and the degree of gene coverage by mutation processes. The following methods are currently used.
The diet is aimed at reducing the absorption of iron in the intestines; nuts, cocoa, soy, and tea are recommended.
The severe form requires regular blood transfusions, red blood cells, and thawed and filtered red blood cells. The effectiveness is temporary, side effects are possible, but the main thing is to save the patient’s life.
The therapy is supplemented by the daily elimination of excess iron through the administration of chelates - special complexes that increase the effect of the medicine. Desferal is prescribed to bind iron. This drug prevents siderosis (a pathological condition caused by deposition of iron in tissues), but does not affect hemoglobin levels.
If a sharp deterioration of the condition occurs, similar to hemolytic crises, glucocorticoids in large doses are indicated.
Splenectomy is possible for children with a large spleen after the age of five. The most optimal age is 8–10 years. After removal of the spleen, a period of improvement begins, but the risk of infection is dangerous.
For transplantation, a donor matching all parameters is required, preferably a close relative.
Symptomatic remedies include hepatoprotective drugs; large doses of ascorbic acid help remove excess iron from the body.
All forms of thalassemia require medications with folic acid and B vitamins. Against the background of an associated infection, folic acid should be used in large doses during pregnancy, since ineffective hematopoiesis in thalassemia significantly increases its consumption by cells.