1.What is sickle cell anemia and its causes
Sickle cell anemia is an inherited blood disorder.
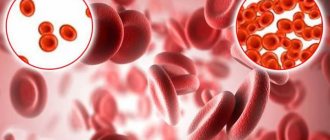
Let's try to figure out what it is. Red blood cells contain hemoglobin, a protein that carries oxygen in the blood. Normally, red blood cells are round in shape and flexible, allowing them to move through small blood vessels to deliver oxygen to all parts of the body.
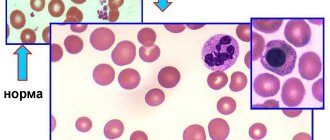
Sickle cell anemia causes blood cells to take the shape of a crescent, or sickle. In this form of blood cells, red blood cells break down very easily, which causes anemia
. Sickle red cells live only 10-20 days instead of 120 days, as is normal for healthy cells. In addition, damaged red blood cells clump and adhere to the walls of blood vessels, blocking blood flow. This can cause severe pain and permanent damage to the brain, heart, lungs, kidneys, liver, bones and spleen.
Causes of sickle cell anemia
Human sickle cell anemia is caused by a genetic abnormality in the hemoglobin gene. This abnormality results in sickle hemoglobin. When oxygen is released from sickle hemoglobin, it sticks together and forms long rods that damage the red blood cell and change its shape. Sickle-shaped red blood cells cause the symptoms of sickle cell anemia.
Human sickle cell anemia is not contagious. This is a genetic disease that a person is born with. The disease develops when a child inherits two abnormal hemoglobin genes, one from each parent. People who have inherited only one abnormal hemoglobin gene are carriers of it, but they will not have anemia or symptoms of the disease.
A must read! Help with treatment and hospitalization!
Sickle cell anemia
Terminology
For more information about the composition and biological role of blood, about the essence of anemia, see the material “Anemia.
Blood and bloodlessness." Sickle cell anemia is a severe hereditary disease, also known as drepanocytosis, meniscocytosis, S-hemoglobin disease, Gerrick's syndrome, African hemolytic anemia (hemolytic - Greek lit. "dissolving, decomposing blood"). The modern history of the study of this type of anemia begins, it is believed, in the middle of the 19th century , when an autopsy revealed no spleen in the body of an African slave executed for escaping. Then in 1910 a report appeared, authored by cardiology professor James B. Herrick and his intern Ernest E. Irons. In a blood test of a 20-year-old student who had been treated since 1904 for “anemia, muscular rheumatism and biliousness” (he died of pneumonia in 1916), Irons observed and first described red blood cells of a strange shape, which he defined as “elongated and sickle-shaped.” " The name "sickle cell anemia" was first used as a diagnosis by Verne Mason in 1922 . Since the 1930s, epidemiological studies of this disease began in samples of children of African descent.
It can be assumed that the genetic mutation leading to sickle cell anemia appeared on the African continent - and, oddly enough, became established in the population as a useful trait: carriers of the gene and patients with this form of anemia show relative resistance to malaria. The malarial plasmodium parasitizes red blood cells and destroys them, however, red blood cells, genetically mutilated and carrying a completely different hemoglobin (see below), are “inedible” for plasmodium. And although the anemia of malaria is not sweeter, the mortality rate of swamp fever is still higher: those who had at least some immunity survived.
In an actively migrating humanity, which is becoming increasingly international and gradually interracial, sickle cell anemia is widespread. However, a certain endemicity remains today: in Equatorial Africa, the Mediterranean basin, India, and the Middle East, the frequency of genetic carriage (not to be confused with the incidence in the clinical form) reaches 40%. According to a special study conducted in 2001 in Jamaica, the average life expectancy of patients with homozygous sickle cell anemia is 53 years for men and 58 years for women. About 90% live to be 20 years old, and about half live to be 50 years old.
In other words, the problem is very serious, it is characterized by high medical and social significance and requires effective solutions, the search for which is currently being carried out by leading specialized research centers. The prevalence of hereditary hemoglobinopathies (including sickle cell anemia) in the world is estimated at 3-7%. In the early 2010s, WHO classified sickle cell disease as a global health problem.
The epidemiological situation and mortality rates in third world countries are unknown or not sufficiently reliable.
According to the literature, due to migration processes, the incidence in Russia shows a significant and stable tendency to increase, especially in Moscow and St. Petersburg.
2. Symptoms of the disease
Symptoms of sickle cell anemia may include:
- Severe pain;
- Anemia;
- Chest pain and difficulty breathing;
- Joint pain, arthritis;
- Blockage of blood flow in the spleen or liver;
- Severe infections.
People with sickle cell disease develop severe pain in the chest, back, arms, legs, and abdomen. Additionally, pain can occur in any part of the body. Sickled red blood cells in the lungs can cause serious illness with symptoms such as chest pain, fever and difficulty breathing. Sickle cell anemia can also cause permanent damage to the brain, heart, kidneys, liver, spleen and bones. The special thing about this disease is that the severity and symptoms of the disease can vary greatly from person to person, even if they are blood relatives.
Visit our Therapy page
Treatment.
Today, sickle cell anemia is an incurable disease. However, its identification is extremely important for the correct treatment of other diseases in these patients, as well as for providing them with adequate surgical and obstetric care. Proper management of patients with chronic sickle cell anemia helps prevent severe exacerbations (crises) and prolong life.
In some cases, the development of a severe crisis can be prevented by the rapid administration of antibiotics in its early stages (to stop the infection) and fluid infusion (hydration). Treatment for a full-blown crisis usually includes oxygen, painkillers, intravenous fluids, and antibiotics. Sometimes it is necessary to resort to transfusions of red blood cells, as well as use many other drugs, such as anticonvulsants, to relieve individual symptoms. See also ANEMIA.
3. Diagnosis of sickle cell anemia
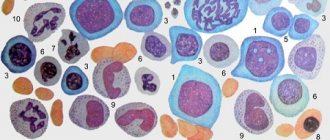
Sickle red blood cells can be seen when a blood sample is examined under a microscope. But to diagnose human sickle cell anemia, a special blood test is used - hemoglobin electrophoresis
, which measures the amount of abnormal hemoglobin. Depending on the amount of sickle hemoglobin measured, it can be determined whether a person is a carrier of the gene or has sickle cell disease.
There are also quick screening tests,
which help identify sickled red blood cells or clumps of abnormal hemoglobin when oxygen leaves the blood. But these tests are rarely used because they cannot distinguish between disease and the simple presence of a disease gene.
Perinatal diagnosis of sickle cell anemia
carried out by studying the DNA of fetal cells obtained through chorionic villus sampling or amniocentesis.
About our clinic Chistye Prudy metro station Medintercom page!
Diagnostic studies.
The diagnosis of sickle cell anemia is based on the analysis of the physical properties of hemoglobin. The first and oldest method of such analysis is the study of the so-called. "wet smear" When a blood smear is moistened with sodium metabisulfite, red blood cells give up oxygen and a characteristic change in their shape can be seen under a microscope. For greater accuracy, the study is repeated after 24 hours. Another, more common method is based on the detection of sickle cell hemoglobin by its reduced solubility in certain buffer solutions, which is determined by the turbidity of the solution containing such hemoglobin. The widespread use of this method is due to the ability to quickly obtain results (within 10–15 minutes).
Unfortunately, these methods do not allow one to distinguish a heterozygous state from a homozygous one. Currently, this can only be done using hemoglobin electrophoresis, i.e. analysis of its mobility in an electric field. Without such an analysis, neither accurate diagnosis nor reliable counseling is possible, but for mass examinations it is too expensive and time-consuming.
Symptoms during the height of the disease
At the height of the disease, hemolytic anemia occurs constantly. In the bone marrow of tubular bones, the medulla grows, which changes the structure of the skeleton. Patients have typical changes:
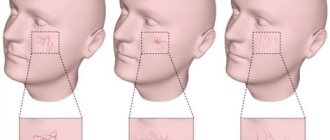
- elongated (“tower”) skull and distinct bulges of the forehead;
- rachiocampsis;
- thin bones.
Compensatory activation of hemoglobin synthesis in the liver and spleen leads to their sharp increase, thrombosis of regional vessels, hemosiderosis (increased iron deposition) in the tissue of the pancreas, heart, and liver. This contributes to the development of cholelithiasis, cirrhosis, and diabetes.
In the kidneys with sickle cell anemia, small arteries of the glomeruli are thrombosed, filtration is reduced, which leads to renal failure with the accumulation of waste and nitrogenous substances.
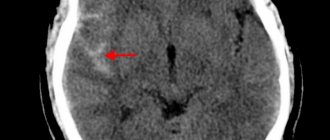
Neurological symptoms are manifested by impaired cerebral circulation (stroke) with paralysis or paresis of muscle groups and cranial nerves.
Trophic changes in the form of non-healing ulcers on the feet and legs associated with thrombosis of the arteries of the legs are typical.
Patients endure this period for up to five years. At the same time, the mortality rate is the highest.
Clinical features in patients of different ages
Doctors encounter different manifestations of sickle cell anemia in children and adults.
During the newborn period - up to three months of age, the child is no different from other children, gaining weight normally. The first symptoms are swelling and pain in the hands and feet. At 6 months, dactylitis (inflammation of individual fingers and toes) often develops. By the age of one year, weakness in the legs, curvature of the legs, and the baby’s reluctance to walk are revealed. The child is pale, the skin has a jaundiced tint.
The clinical picture is associated with thrombosis of small capillaries and impaired blood flow in the extremities. Blood clots resolve on their own, but help is needed to relieve pain. Serious complications are caused by infection. The risk of sepsis and death of the child increases sharply.
Inflammation of the digital phalanges is more common in children
After 5 years and at school age, the likelihood of sepsis is much lower. Because it has already developed its own protective immunity and antibodies to some bacteria. The problems are associated with periodic pain in the hands. In adolescence, attention is drawn to the short stature of children and delayed development of secondary sexual characteristics. Over time, they are compared with other young people.
Women with sickle cell anemia can give birth to a child, but pregnancy will require special monitoring by a hematologist and additional prevention of complications.
In adults, thrombotic manifestations are observed in the kidneys (development of renal pathology), in the retina of the eyes (visual impairment to complete blindness), in the brain (stroke), and pulmonary infarction pneumonia.
All symptoms rarely occur in one patient. Usually there is one variant of pathological changes.
Clinical manifestations of the initial period
Symptoms of sickle cell anemia are more pronounced when there is a high level of hemoglobin S in the blood. They manifest themselves through two mechanisms:
- increased thrombus formation in small vessels;
- manifestations of hemolysis, destruction of red blood cells.
In the initial period of the disease, the bone marrow hematopoietic system and arteries feeding the bones are affected. Symptoms appear:
- pain in the bones is first aching, then very intense, requiring the administration of analgesics;
- swelling of the legs, joints;
- increased patient anxiety;
- infection can lead to osteomyelitis.
Sickle cell anemia occurs in the form of crises, they are called hemolytic. Provoked by any acute infection. The inability to quickly restore red blood cell synthesis leads to death.
Other options:
- sequestration (storage) of blood in the liver and spleen, in the absence of hemolysis, abdominal pain appears, a sharp increase in these organs, a typical drop in blood pressure;
- Pulmonary thrombosis causes pulmonary infarction if the clotting disorder occurs at the level of the pulmonary vessels.
How do altered properties of hemoglobin affect hematopoiesis?
Hemoglobin S forms crystalline chains that change the shape of the red blood cell to a sickle shape, making it look like a crescent. In this form it:
- loses the ability to bind an oxygen molecule and transport it through the bloodstream;
- undergoes increased breakdown in the spleen, since the “life” of cells is shortened;
- not resistant to hemolysis (dissolves in the blood under the action of enzymes).
Impaired transportation is manifested by tissue hypoxia of internal organs. The bone marrow constantly receives “requests” for the synthesis of normal globin, so the main stem cells from which leukocytes and platelets develop are injured.
Various types of hemolytic crises
Hemolytic crisis conditions differ in the leading symptom, so it is customary to distinguish types:
- thrombotic - leading to vascular thrombosis, organ infarctions;
- hemolytic - the development of hemolysis, jaundice, a decrease in hematocrit in the blood, an increase in bilirubin prevails;
- aplastic - against the background of a threatening drop in hematocrit, the infection causes inhibition of bone marrow hematopoiesis, increased consumption of folic acid;
- sequestration - in the blood there is a general inhibition of cell growth, a sharp increase and soreness of the liver and spleen.
A rarer type is acute chest syndrome. It is characterized by a sudden increase in body temperature, chest pain, and shortness of breath. An x-ray reveals multiple infiltrates in the lungs.
The duration of crises can be up to a month.
Is it possible to prevent the disease?
There is no way to prevent chromosome damage. Spouses who know that there are cases of the disease in the family should seek genetic advice about their upcoming pregnancy. The calculated risk of producing a homozygous heir calls into question the possibility of having a child.
At the birth of a heterozygous baby it is necessary:
- provide him with proper nutrition with sufficient folic acid;
- monitor increased fluid intake;
- protect from infections.
All initial manifestations must be treated urgently. Then a milder course of sickle cell anemia is possible.