A biochemical blood test is a laboratory diagnostic method used in medicine, the results of which make it possible to assess the functional state of organs and systems of the human body. With its help, you can determine the presence of an active inflammatory or rheumatic process in a patient, determine the function of the kidneys, liver, as well as an imbalance of microelements and a violation of water-salt metabolism. A biochemical blood test allows you to correctly establish a diagnosis, prescribe adequate treatment and, if necessary, adjust it and determine the stage of the disease.
Blood for a biochemical blood test is taken on an empty stomach from a vein. Before the examination, it is forbidden to drink (except water), eat, chew gum and take any medications 6-12 hours before the procedure. Sometimes there may be exceptions when it is extremely important to take the medicine early in the morning. In such cases, be sure to consult with your doctor, he will be able to give more precise recommendations. The reliability and accuracy of the results depends on whether the patient was properly prepared for the analysis and whether he followed all the doctor’s recommendations. Do not forget that a biochemical blood test is performed strictly on an empty stomach in the morning. The lead time for such analysis is from 1 day.
Standards for biochemical analysis in children
The standards for biochemical analysis in children change as they grow. After all, your baby is growing, and his organs and systems are developing and improving. That is why each age group has its own biochemical blood test norms. As a rule, diagnostic laboratories indicate reference (normal) values taking into account age in a separate column on the analysis result form. But, if your laboratory has not written the norms, or indicated the norms without taking into account the child’s age, do not be alarmed. Be sure to consult with your personal pediatrician who knows the norms of biochemical blood tests and the individual characteristics of your baby.
Boys and girls
Due to physiological characteristics, collecting urine from boys is much easier. You can “catch” the stream of urine with a container or lower your penis into the container and wait. It is very convenient to use a urinal, which is made specifically for boys. Both the penis and testicles are placed in such a urinal.
In little girls, collecting urine is much more difficult. If there is a urinal, then you need to attach it in a circle, capturing only the labia majora. If there is no urine bag, then you will have to use a wide, but not very deep plate. Pre-wash it and pour boiling water over it. Hold the girl over (or next to) a plate and wait for her to pee.
Who can you trust to decipher a biochemical blood test?
Many parents believe that decoding the biochemical analysis of a child’s blood consists only of comparing the age-related norms with the results of the child’s analysis. And, therefore, out of ignorance, they entrust the decoding of the analysis to Internet resources or special tables. This way of deciphering a child's analysis is erroneous and very dangerous.
A pediatrician should decipher the biochemical analysis of a child’s blood. After all, only a doctor knows the child’s complaints, the peculiarities of the mother’s pregnancy and childbirth, the characteristics of the baby’s growth and development from the first day of life, and can conduct an examination and identify the child’s hereditary predisposition. The pediatrician compares all this data with the results of a biochemical analysis of the child’s blood and makes a transcript.
You can make an appointment with a doctor at the Center through:
online appointment form
or by calling the Center in Moscow:
+7,, +7 (962) 947-38-08
during the working hours of the Center (daily, seven days a week and holidays, from 9.00 to 21.00)
PRICE LIST for medical services of the Center | |
| TYPES OF MEDICAL SERVICES | Cost, rub. |
| ** The main types of analyzes are given. It is possible to conduct other types of analyzes - please check with the Center administrators for details. | |
| Potassium | 400 |
| Total calcium | 290 |
| Magnesium | 770 |
| K/Na/Cl | 400 |
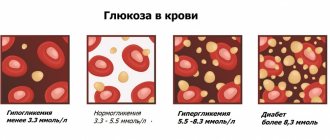
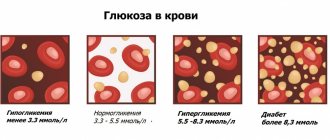
| Glucose (blood sugar test) | 280 |
| Phosphorus | 300 |
| Sodium | 770 |
| Rheumatoid factor | 550 |
| C-reactive protein | 550 |
| Bilirubin and its fractions (total, direct and indirect) | 540 |
| Lactate dehydrogenase (LDH) | 280 |
| Alkaline phosphatase | 280 |
| Gamma-GT | 280 |
| Urea | 280 |
| Creatinine | 280 |
| Ionized calcium | 480 |
| Total protein | 290 |
| Total bilirubin | 290 |
| Alamine transaminase (ALT) | 280 |
| Aspartate transaminase (AST) | 280 |
| Laboratory visit within the Moscow Ring Road | 1 720 |
| Alpha-aminase | 370 |
| Emergency laboratory visit within the Moscow Ring Road | 1720 |
| Laboratory visit outside the Moscow Ring Road | 2300 |
| Cholesterol (HDL) | 340 |
| Cholesterol (LDL) | 300 |
| Total cholesterol | 300 |
Coprogram
You will need a clean container to collect this test. You can buy it at the pharmacy, it is almost no different from a container for collecting urine, the only thing is that it has a small spoon attached to its lid. It is not necessary to wash the child immediately before collecting this analysis.
The easiest way to collect feces is from a diaper - you need to take the contents from the surface. The volume of collected material should be approximately 5–10 g (1–2 teaspoons). If the baby's stool is liquid and analysis is necessary, you will have to place the child on medical oilcloth and then carefully pour the contents into a container.
For dysbacteriosis, feces cannot be collected from a diaper.
Enterobiasis: Collection of biomaterial is carried out only in the morning, before 10.00 am. Do not wash your child either the evening before or in the morning.
Prepare a glass slide by wiping it dry. Place the child on the side, bend the legs at the knees and bring them to the stomach, spread the buttocks and collect scrapings from the perianal folds around the anus using the “print” method in several places with adhesive transparent tape (adhesive tape) 4-5 cm long. Carefully stick the adhesive tape on a glass slide sticky side down. Bring it to the laboratory in this form. The slide can be obtained from the treatment room.
Symptoms
Type I The first signs in a child appear between the ages of 2 months and six months.
- Apathy or, conversely, increased irritability.
- Lack of interest in the environment, people, objects, settings.
- Frequent regurgitation.
- Allergic dermatitis.
- Violation of muscle tone.
- Low pressure.
- Cramps.
- Sometimes microcephaly (small size of the skull relative to other parts of the body) and hydrocephalus (excess fluid surrounding the brain) develop.
Characteristic symptoms include hypopigmentation of the skin, hair, and iris. Urine has a specific musty smell or is also called a “mouse” smell. Epileptic seizures are observed in half of the patients and are often the first pronounced clinical symptom. The attack is characterized by “salaam” convulsions, reminiscent of nodding. They occur frequently and do not respond well to anticonvulsant treatment.
If PA concentrations are not adjusted, the disease progresses. As a rule, the IQ level in such children does not exceed 20, with the norm being 85. Mental retardation is so severe that there are no emotional reactions, psychopathy and schizophrenia-like disorders are observed.
Type II The first symptoms appear in the first year of life.
- Increased excitability.
- Developmental delay.
- Profuse drooling.
- Reduced blood pressure.
- Frequent increase in body temperature.
- Tendon hyperreflexia (increased reflexes) or spastic tetraparesis (weakness of all four limbs).
- Myoclonic epilepsy (generalized seizures, predominantly occurring upon awakening).
- Microcephaly.
A distinctive feature of the second type is the death of neurons, disruption of folate metabolism, as well as calcification in various parts of the brain. The disease progresses rapidly and can lead to the death of a child within 2 to 3 years.
III type. Symptoms of pyruvoyltetrahydropterin synthetase deficiency are similar to those of Parkinson's disease:
- Postural instability and gait difficulties. It is difficult or impossible to maintain a certain position of the whole body or limbs.
- Hypokinesia (low motor activity, limited tempo and range of movements).
- Hypersalivation (increased salivation).
- Swallowing disorders.
- Oculogyric crisis (symmetrical deviation of both eyes, usually directed upward).
In 80% of cases, this type of disease is accompanied by a decrease in the amount of biogenic amines in the cerebrospinal fluid. Treatment is complicated by the fact that an early decrease in FA concentration can cause serious pathological changes. Failure to comply with diet therapy will lead to slower speech development, low intelligence, and memory problems.
Classification
Phenylketonuria currently does not have a generally accepted worldwide classification. There is ongoing debate over this issue, as well as research into the disease. A little earlier, before deciphering genes, it was believed that the degree of damage to intellectual abilities depended on the degree of enzyme activity. Therefore, the current qualification is considered outdated. It also does not take into account other symptomatic factors.
When diagnosing they put:
- Type I (PAH deficiency) - FA concentration is more than 20 mg/dl.
- The average form of PKU is FA from 8.1 to 20 mg/dl.
- Mild form of HPA level - FA from 2.1 to 8.0 mg/dl.
At levels up to 8.0 mg/dL, phenylketonuria is considered benign. It does not require special treatment, but observation is necessary for the first year of the child’s life. The condition is monitored by a pediatrician, neurologist, and geneticist.
There is also another form of phenylketonuria that does not require correction. This is a transient form of HPA during the neonatal period. Occurs, as a rule, with prematurity, which is due to the functional immaturity of the body. Transient phenylketonuria is a temporary increase in FA levels that can rise to critical values. In this case, there is no clinical manifestations or the manifestations are very minor. After a few months, biochemical parameters returned to normal.
Pathogenesis
The mechanism of the origin and development of phenylketonuria is associated with a violation of the metabolism of an organic compound - the amino acid phenylalanine. The metabolic block prevents the conversion of phenylalanine to tyrosine. The amino acid not only is not converted, but accumulates in the form of toxic metabolites:
- phenyllactic acid;
- phenylpyruvic acid;
- phenylacetic acid;
- phenylethylamine and so on.
The accumulation of phenyl substances has a toxic effect on the central nervous system. At the moment, the mechanism has not yet been fully studied; doctors do not know the pathogenesis of brain dysfunction.
There are suggestions that damage to the nervous system is the result of a number of factors. Among them are both the direct toxic effects of phenylalanine and disruption of the metabolism of proteins, lipoproteins and glycoproteins, failure of homonal metabolism and membrane transport of amino acids. All this together is important for the maturation and proper functioning of the central nervous system.
What not to do
- There is no need to squeeze the described diaper into a container and submit such an analysis to the laboratory: the results will be unreliable, because various microorganisms and tissue fibers will get from the diaper into the contents of the container.
- There is no need to use homemade urine bags. For example, making them from a plastic bag is both inconvenient and unhygienic.
- You cannot freeze the container with the analysis (any kind). The material collected the day before is unsuitable for research.
- Enemas and laxative suppositories should not be used to stimulate bowel movements, otherwise the laboratory material will contain foreign impurities and the study will be unreliable.
When the baby grows up, the procedure for collecting tests will become much simpler. Both urine and feces can be collected from the potty. The main thing is that before collecting samples, the pot must be washed clean, but without using cleaning products. Just wash it with baby soap or baking soda and rinse thoroughly with water.
Treatment
Symptomatic therapy for any form of phenylketonuria is ineffective. There is only one way to prevent the negative consequences of the disease - diet therapy. High-protein foods and phenylalanine-containing foods are excluded from the diet. The missing amount of protein is replenished with specialized therapeutic nutrition, with the lowest possible content of the amino acid FA or completely devoid of it. It should be taken into account that the effectiveness of therapy directly depends on the time the correction began and the pathological changes that have already occurred.
The goal of therapeutic nutrition in the classic form of the disease is to prevent the development of disorders of the central nervous system, physical and mental development. A mild form of HFA allows for an expansion of the diet under the strict supervision of a doctor over the child’s condition and biochemical parameters. Prohibited: meat, fish, nuts, chocolate and legumes, all types of eggs, cottage cheese and cheeses. You should also avoid foods that contain the artificial sweetener aspartame.
The criterion for the effectiveness of treatment is the level of FA in the blood.
Diagnostics
Phenylketonuria can be detected in the first days after birth before any symptoms appear. To determine the concentration of phenylamine in the blood, carry out:
- microbiological test;
- chromatography;
- fluorimetry;
- mass spectrometry.
In all cases, the biological material is dry spots of capillary blood from the baby.
Recently, testing for phenylketonuria has been included in the neonatal screening program. It is carried out on all newborns; the study is especially important for premature babies. The diagnostic criterion is an increased concentration of phenylalanine at a rate of 0 - 2 mg/dl. An elevated level requires further diagnostic testing. It will be necessary to establish the very fact of the presence of phenylketonuria and identify its cause.
- If the screening test shows high PA level results, the following may additionally be prescribed:
- Phenylalanine load diagnostics to identify the nosological form of the disease.
- Molecular genetic analysis to determine the form: classical, type II or III.
- Sequencing of the RAS gene, if molecular genetic diagnostics gave a negative result for the PAH gene.
- Analysis for pterins in urine to exclude pterin-dependent forms.
Differential diagnosis of phenylketonuria is carried out with such pathologies as liver dysfunction, galactosemia and other diseases.