Thrombocytopenia in children and adults can be an independent disease (primary form) or a symptom of pathologies of other systems and organs (secondary thrombocytopenia). The problem most often occurs in preschoolers and people over 40 years of age.
According to the criterion of duration of occurrence in the body, the disease is classified into:
- Spicy. There are no immediate symptoms; thrombocytopenia affects the organs for no more than six months.
- Chronic. The decrease in platelets in the blood continues for more than six months. Treatment is long-term, taking up to two years.
Causes
Platelets are formed elements of blood, nuclear-free flat plates ranging in size from 1 to 2 microns. They are formed from megakaryocytes (precursor cells) in the red bone marrow.
Megakaryocytes are relatively large cells. They have long processes and are almost completely filled with cytoplasm. During maturation, tiny fragments of their processes break off and enter the bloodstream. These are platelets. According to scientific data, from 2000 to 8000 platelets can be formed from one donor cell.
Thrombopoietin, a protein hormone produced in the kidneys, liver and muscles, is responsible for the development and growth of megakaryocytes. It is transported through the bloodstream to the red bone marrow, where it ensures the formation of megakaryocytes and platelets. The more platelets there are, the more the process of thrombopoietin formation is inhibited. This allows you to maintain the number of blood cells at the same level.
If any of these steps fail, the number of platelets in the peripheral blood decreases. Thrombocytopenia develops.
Taking into account the causes and mechanism of development, the disease comes in different forms:
- hereditary;
- productive;
- destruction;
- consumption;
- redistribution;
- breeding.
Hereditary thrombocytopenias
Hereditary thrombocytopenia is observed with various congenital anomalies. Its causes are genetic mutations:
- Wiskott-Aldrich syndrome. Caused by mutations that cause the red bone marrow to produce very small platelets (less than 1 µm in diameter). Due to their abnormal structure, they are quickly (within a few hours) destroyed in the spleen.
- May-Hegglin anomaly. A very rare genetic pathology, due to which the process of separating platelets from megakaryocytes is disrupted. As a result, the number of blood cells decreases, but their sizes become gigantic (6-7 microns). In parallel, there is a disturbance in the formation of leukocytes (leukopenia).
- Congenital amegakaryocytic thrombocytopenia. Typically, this form of thrombocytopenia is diagnosed in infants. It is associated with impaired platelet production by the bone marrow.
- Bernard-Soulier syndrome. Thrombocytopenia in a child appears only if he has inherited a defective gene from both his mother and father. The disease manifests itself in early childhood. It is characterized by the formation of functionally incompetent giant platelets (6-8 µm), unable to attach to damaged vascular walls and communicate with each other.
- TAR syndrome. A very rare cause of congenital thrombocytopenia, combined with the absence of both radius bones.
Productive thrombocytopenia
The group of productive thrombocytopenias includes pathologies of the hematopoietic system associated with disruption of the process of platelet formation in the red bone marrow. Causes of diseases:
- Acute leukemia. Stem cells mutate, and a large number of clones appear that are unable to perform specific functions. Gradually, clones displace hematopoietic ones from the red bone marrow. Not only the number of platelets decreases, but also the number of leukocytes, lymphocytes and erythrocytes.
- Myelodysplastic syndrome. The bone marrow cannot produce enough healthy cells. Hematopoietic cells multiply rapidly, but their maturation processes are disrupted. As a result, many functionally immature cells are formed (among them platelets) that undergo apoptosis (self-destruction).
- Aplastic anemia (the body produces very few new blood cells).
- Cancer metastases. Tumor cells in the final stages of cancer begin to leave the primary focus and spread throughout the body. Settling on tissues and organs, they begin to actively reproduce. This leads to the displacement of hematopoietic cells from the red bone marrow.
- Myelofibrosis. The stem cells mutate and the bone marrow is replaced by scar tissue. Foci of hematopoiesis develop in the liver and spleen (the size of the organs increases greatly due to this).
- Radiation. Ionizing radiation has a destructive effect on the hematopoietic cells of the red bone marrow. They begin to mutate.
- Alcoholism. Alcohol inhibits the processes of hematopoiesis in the red bone marrow, which reduces the content of platelets, leukocytes and red blood cells in the blood.
- Cytostatic drugs. Used to treat tumors. They can inhibit hematopoiesis in the bone marrow, which reduces the number of platelets.
- Allergic reactions to certain medications (diuretics, antibiotics, antipsychotics, anticonvulsants and anti-inflammatory drugs, antidiabetic drugs).
- Megaloblastic anemia. They develop with a deficiency of folic acid and vitamin B12.
Thrombocytopenia destruction
The cause of thrombocytopenia in this case is the increased destruction of platelets in the spleen (can also be in the lymph nodes, vascular bed or liver). Pathology is observed when:
- thrombocytopenia of newborns;
- Evans-Fisher syndrome;
- some viral diseases (viral thrombocytopenia);
- post-transfusion thrombocytopenia;
- taking medications (drug-induced thrombocytopenia);
- idiopathic thrombocytopenic purpura.
Thrombocytopenia of newborns
Occurs if there are antigens on the surface of children's platelets that are not on maternal platelets. Antibodies produced by the mother's body enter the newborn's bloodstream and destroy his platelets. The described process can last up to 20 weeks of intrauterine development. As a result, the child may be born with thrombocytopenia.
Fisher syndrome
It is a consequence of systemic diseases - autoimmune hepatitis, rheumatoid arthritis, systemic lupus erythematosus. Also, Evans-Fisher syndrome can develop against a background of relative well-being. Then they talk about idiopathic thrombocypenia.
Viral thrombocypenia
Once entering the body, viruses actively multiply. Antibodies are formed on the surface of the affected cells, and the cell's own antigens also change. The affected cells are destroyed in the spleen. Among the viruses that can cause thrombocypenia are measles, rubella, influenza, and chickenpox.
Post-transfusion thrombocytopenia
Post-transfusion thrombocytopenia is a consequence of platelet or blood transfusion. They are characterized by pronounced destruction of platelets in the spleen. The cause of the disease is that foreign platelets enter the body during a transfusion, to which antibodies begin to be produced.
Drug-induced thrombocytopenia
Some medications can bind to antigens on the surface of blood cells. Antibodies are produced to the formed complexes, which causes platelets to begin to be destroyed in the spleen. Among the “provoking” drugs are Quinidine, Meprobamate, Chloroquine, Ranitidine, Heparin, Cimetidine, Gentamicin, Ampicillin, etc.
Idiopathic thrombocytopenic purpura
Idiopathic thrombocytopenic purpura (also known as autoimmune thrombocytopenia or essential thrombocytopenia) is characterized by a sharp decrease in the level of platelets in the peripheral blood. At the same time, the composition of other blood elements is not disturbed.
Idiopathic thrombocytopenic purpura: from Werlhof to the present day
Spontaneous skin bleeding, known for more than 2500 years, was already called “purpura” in the Greek and Roman healing period. In 1735, Paul Werlhof (ITP is also called "Werlhof's disease") described a 16-year-old girl with epistaxis and mucosal bleeding that was controlled by the use of citric acid. He named this disease "Morbus Maculosus Haemorhhagicus". But noticeable progress in the treatment of patients with ITP was achieved later: in 1916 in Prague, Professor Schloffer removed the spleen from a woman with this disease. A significant increase in platelet counts followed surgery. And until now, splenectomy is one of the treatment options for patients with ITP. However, the most complete picture of this pathology, our understanding of the mechanisms of its occurrence and approaches to diagnosis and therapy have begun to emerge only recently.
Idiopathic thrombocytopenic purpura (ITP), or primary immune thrombocytopenia (ITP), is an acquired autoimmune disease characterized by isolated thrombocytopenia with a platelet count below 100x109/L. It can manifest itself as a hemorrhagic symptom of varying severity - from petechial skin hemorrhages to life-threatening bleeding. Both children and adults get sick. The etiology of ITP is unknown. That's why it's called "idiopathic". Among the triggering factors, the largest group includes infections, pregnancy, as well as vaccinations and stress.
It is known that the dominant mechanism for the development of thrombocytopenia in ITP is due to the production of autoantibodies to the membrane structures of platelets and their precursors - megakaryocytes, which lead to increased destruction of platelets by phagocytes, mainly in the spleen, less often in the liver, and insufficient production of platelets in the bone marrow. Patients with ITP develop primarily IgG autoantibodies against platelet surface glycoproteins GPIIb/IIIa or GPIb/IX. The process of forming an immune response to one’s own platelets is complex, multi-stage, and cyclical. B lymphocytes, T lymphocytes, NK cells, and macrophages take part in it. In addition to antibody formation, subpopulations of T-lymphocytes and the development of an imbalance in the T-cell component of the immune response play a major role in the pathogenesis of ITP. An association has been identified between ITP and some candidate genes, which also indicates the presence of a genetic predisposition to ITP.
Taking into account the concomitant pathology, the patient develops a certain phenotype of the disease. Thus, the pathogenesis of ITP is associated with profound disorders of the immune system. In this regard, idiopathic thrombocytopenic purpura has been renamed to primary immune thrombocytopenia of unknown etiology. Accordingly, in all other forms, immune thrombocytopenia with a known etiology will be a symptom of other autoimmune diseases - systemic lupus erythematosus (SLE), antiphospholipid syndrome (AFLS), rheumatoid arthritis (RA), etc.
Currently, the search continues for etiopathogenetic mechanisms of the development of this rare pathology, which could stratify patients into risk groups to individualize treatment tactics.
In the literature, ITP is described as a rare orphan disease. It must be said that in the medical world there is no single definition of this group of diseases. In some countries, orphan pathologies are identified depending on the number of sufferers, in others - on the availability of treatment methods, in others - only chronic, life-threatening ones are classified as rare diseases.
Russia has its own history of orphan diseases. Since the time in our country the definition of “orphan diseases” was legislatively adopted (Law No. 323 “On the fundamentals of protecting the health of citizens in the Russian Federation” dated November 21, 2011)1, namely: rare (orphan) diseases are diseases that are widespread no more than 10 cases of the disease per 100 thousand population, all oncohematological and many hematological diseases began to be considered orphan. As for ITP, according to Decree of the Government of the Russian Federation No. 403 of April 26, 2012, idiopathic thrombocytopenic purpura (D69.3) was included in the short list of life-threatening and chronic progressive rare (orphan) diseases leading to a reduction in the life expectancy of citizens or their disability. This short list also included such hematological conditions as paroxysmal nocturnal hemoglobinuria (Marchiafava-Miceli disease), aplastic anemia, hereditary metabolic diseases, hemolytic-uremic syndrome, etc.
All these legislative decisions led to the creation of a department - a hospital for orphan diseases (headed by Professor E.A. Lukina) at the Federal State Budgetary Institution "National Medical Research Center of Hematology" in 2012. The nosological range of pathologies dealt with by the department is very wide.
Medicines developed to treat rare diseases are also called orphan drugs and include a list of expensive drugs. Assigning orphan status to diseases and any drugs is a social and political issue in many countries, as well as in Russia. Government support for rare disease research has led to medical breakthroughs that could not have been achieved under the previously existing funding system.
The orphan disease status for ITP also opened up new opportunities for improving its diagnosis and treatment with modern methods that could not be achieved without it. We are talking primarily about two orphan expensive thrombopoietin receptor agonist (aTPO) drugs (romiplostim from Novartis and eltrombopag from Amgen). Medicines are provided at the expense of the budgets of the constituent entities of the Russian Federation.
Epidemiology
It should be recalled that in our country, the incidence of ITP at the population level was not studied until 2014. And there was not enough information to assess the course, effectiveness and safety of various treatment options for patients with ITP. To solve these problems, under the auspices of the National Hematological Society (chairman of the Supervisory Board of the NGO - chief freelance hematologist of the Russian Federation, director of the Federal State Budgetary Institution "National Medical Research Center for Hematology" of the Ministry of Health of Russia, academician of the Russian Academy of Sciences, Professor V.G. Savchenko) in early December 2012, " Registry of diseases of the blood system.”3 Since 2014, work has started in its subsection “ITP” - a multicenter prospective observational cohort study “Epidemiological and clinical characteristics of ITP in adults in Russia” (head: A.L. Melikyan) began.
According to the register of the National Hematological Society (NGO), the incidence of ITP in the adult population in the Russian Federation averages 2.0 (1.6‒3.6) per 100 thousand population per year. ITP has no geographical features. Men get sick 2–3 times less often than women. The largest proportion of patients (45.4%) were in the age group from 18 to 40 years, in the group from 41 to 60 years - 26.0% and over 60 years - 28.6%. Thus, among patients with ITP, 71.4% are of working age. The highest incidence of ITP was registered in women of fertile age 4. Our results are quite comparable with data from registers in other European countries.
Diagnostics
The main clinical manifestation of ITP is hemorrhagic syndrome, and the prognosis of the disease depends entirely on its severity. The risk of bleeding in patients with ITP is assessed by the platelet count in a peripheral blood test. According to the register, in 70.0% of cases, the number of platelets at the onset of the disease ranges from 3 to 30x109/l, among them, 35% have a critical level of platelets (from 3 to 10x109/l) with the risk of developing spontaneous alarming, life-threatening bleeding, which requires immediate treatment.
Hemorrhagic syndrome manifests itself in the form of: skin hemorrhages - 77% of cases; bleeding of oral mucosa – 39%; nosebleeds – 31%; menometrorrhagia – 15% (among women); gastrointestinal bleeding – 7%; hematuria – 4%; intracerebral bleeding - 0.9%, others - 1% (retinal hemorrhage, hemorrhoidal bleeding).4
Thus, about 1/3 of patients at the time of diagnosis have hemorrhagic manifestations corresponding to a severe form of ITP (grade 3–4 bleeding according to the WHO classification). ITP is not a genetic disease, but usually accompanies the patient throughout his life and is incurable. The course of the disease is further complicated by the fact that in 60–70% of patients after 12 months (chronic phase), the disease becomes chronic and relapsing again.
ITP is not a genetic disease, but usually accompanies the patient throughout his life and is incurable. The course of the disease is further complicated by the fact that in 60–70% of patients after 12 months (chronic phase), the disease becomes chronic, relapsing, and hemorrhagic syndrome reappears, requiring an anti-relapse course of therapy.
The diagnosis of ITP is a diagnosis of exclusion, i.e. To date, there is no specific test for the disease. Thrombocytopenia of various origins is recorded in a wide range of diseases of hematological, non-hematological and congenital nature, in which isolated thrombocytopenia may be the dominant clinical symptom for a long time. Therefore, to establish the true causes of thrombocytopenia, it is necessary to conduct an expanded diagnostic search at the onset of the disease.4
The initial approach to diagnosing the causes of thrombocytopenia is based on the patient's medical history (his underlying diseases and previous drug therapy), his objective physical examination and examination according to the protocol. The protocol for the differential diagnosis of thrombocytopenia that we developed is included in the National Clinical Guidelines for ITP.5 The most important thing is that all the proposed laboratory and instrumental studies exist in routine practice and are mandatory for all patients with suspected ITP.
After excluding other causes of thrombocytopenia, the diagnosis of ITP is made based on the following criteria:
- isolated thrombocytopenia less than 100.0x109/l, recorded in at least two consecutive blood tests;
- absence of morphological and functional platelet abnormalities;
- absence of pathology of lymphocytes, granulocytes and erythrocytes;
- normal hemoglobin, red blood cells and reticulocytes, if there was no significant blood loss;
- increased or normal number of megakaryocytes in the myelogram;
- normal size of the spleen.
It is important to keep in mind: to quickly relieve hemorrhagic syndrome, patients are often prescribed corticosteroids without examination according to the protocol, which blurs the true clinical picture of secondary immune thrombocytopenia and affects the true results of immunological tests. According to our department, in up to 15–20% of cases, during repeated examination according to the protocol, the diagnosis of ITP is replaced by another. The picture of the disease may change over time; therefore, it is necessary to constantly update data on the patient’s condition and carry out differential diagnosis at each stage of observation/therapy for ITP. Thus, it is very important to carry out a differential diagnosis between primary and secondary thrombocytopenia not only at the onset of the disease, but also during relapse of thrombocytopenia.
Establishing the true causes of thrombocytopenia is extremely important for choosing adequate therapy for such patients
In many cases, patients with primary and secondary ITP receive similar treatment. However, if ITP develops in the context of an underlying disease (eg, SLE, APS, HCV infection, HIV infection, or lymphoproliferative disorder), then treatment should be directed primarily at it.
It should be noted that our doctors’ awareness of this disease is at a fairly high level. Firstly, as stated above, ITP was described 275 years ago. Secondly, ITP is a fairly common rare disease. Every year, more than 300 patients diagnosed with or suspected of ITP are consulted in our department alone. And finally, for the first time in Russia (in 2014), National Clinical Recommendations (NCR) for the diagnosis and treatment of idiopathic thrombocytopenic purpura in adults, along with other nosologies, were developed on the initiative of the Russian Ministry of Health.4 These recommendations are constantly updated and are publicly available.4 The latest edition was published in 2021.
In 2021, updated clinical guidelines from the American Association of Hematology (ASH) and updated International Consensus on the Diagnosis and Treatment of Primary Immune Thrombocytopenia were released. We analyzed these recommendations and compared them with Russian clinical guidelines for ITP.
I want to say that we have not noticed any global changes: special attention is paid to correct diagnosis, and approaches to treatment are described in more detail.
In addition, the study of ITP is included in the educational and scientific plans of our Hematology Center for hematologists, residents, students in advanced training cycles of the Federal State Budgetary Institution “National Medical Research Center for Hematology” of the Ministry of Health of Russia and for specialists in other specialties, since there are hematological masks for various diseases. Our Center organizes on-the-job training for medical specialists on current issues in hematology for both hematologists and other specialists. All these programs are included in the system of continuing medical education. In the era of digitalization and distance learning technologies, holding webinars, master classes, and discussing specific clinical examples, the audience of students is significantly expanding, which contributes to the continuous professional growth of doctors.
Treatment
Splenectomy (SE), introduced into treatment practice at the beginning of the last century by Schloffer, is still one of the treatment options for patients with ITP. It is performed relatively frequently, but over the past three years the number of such interventions has decreased from 26 to 17%, while the proportion of patients receiving modern drugs (thrombopoietin receptor agonists, aTPO) has increased from 5.9 to 45.7%. The same trend is observed abroad, where the frequency of splenectomy is lower than in the Russian Federation.
Since 1951, corticosteroids have been used in the treatment of the disease; they still remain the first line of treatment for patients with newly diagnosed ITP in foreign and Russian protocols - with hemorrhagic syndrome and platelets less than 30‒50.0x109/l or in the absence of hemorrhagic syndrome with thrombocytopenia 9/ l.
According to our registry data, in 92.2% of cases, as a first line, patients receive treatment with corticosteroids, both in the form of standard treatment and pulse therapy, with an effectiveness of up to 70–80% with rapid relief of hemorrhagic syndrome and an increase in platelet counts above a safe level. However, after discontinuation of the drug, a relapse of the disease quickly occurs. Corticosteroids are effective, but they are associated with a large number of potential complications: diabetes mellitus; severe forms of arterial hypertension and arrhythmias; gastrointestinal ulcer, active infections; mental disorders. Therefore, repeated and frequent courses are undesirable. All clinical guidelines strictly limit the duration of treatment with corticosteroids to 3–4 weeks. Unfortunately, due to availability, corticosteroids are also prescribed in subsequent lines of therapy.
In 1980, at the University Children's Hospital in Bern, a 12-year-old boy with acute ITP and immunodeficiency was treated with intravenous immunoglobulin (IVIG), which resulted in a marked increase in platelet count within 24 hours. Since then, IVIG has been widely and successfully used in both first and subsequent lines of therapy as an “ambulance” in emergency and life-threatening situations and is an absolutely invaluable tool for the treatment of pregnant women with ITP. IVIG as a first-line therapy is effective in 80% of cases, the hemostatic effect occurs on days 1–2, and the duration of response is 1–4 weeks. Thus, in fact, after first line therapy, almost all patients are candidates for second line therapy.
To systematize the procedure for prescribing treatment options, the international working group for the study of ITP identified 3 stages of the disease:
- newly diagnosed with a duration of up to 3 months from the moment of diagnosis;
- persistent with a duration of 3–12 months;
- chronic with a duration of more than 12 months.
And the sequence of prescribing therapy for ITP, developed on the basis of many years of clinical experience, is called lines of therapy, which generally correspond to the stages of the disease.
At the end of 2000, without a doubt, a new era of treatment for ITP begins: with modern-generation drugs - thrombocytopoiesis stimulants, thrombopoietin receptor agonists, and TPOs - romiplostim (Novartis) and eltrombopag (Amgen). In 2009, they were approved in Russia as orphan drugs for adults with refractory chronic ITP with or without splenectomy. In 2015, both drugs were included in the List of vital and essential drugs for medical use, approved by order of the Government of the Russian Federation. The use of aTPO (these data are based on evidence-based medicine and are supported by several solid and very high-quality prospective control studies) is effective both before and after splenectomy. With the advent of these drugs, the prognosis of the disease has improved, since they prevent the development of severe side effects of treatment and allow achieving an 80% level of immediate effect.1,6
I believe (like many of my colleagues) that their important features are organ-preserving and corticosteroid-restraining effects.
Another drug for the treatment of ITP, rituximab, has recently appeared in clinical practice, which was developed for the treatment of hematological malignancies. Rituximab is currently used to treat patients with ITP who are refractory to other treatments. Its use in chronic ITP is based on the removal of autoreactive B lymphocytes. Rituximab is included as 3rd line therapy. There is approximately a 60% chance of obtaining a primary response. But in Russia it is not registered for the treatment of ITP, so the decision is made individually by a medical commission.
In 2021, the US Food and Drug Administration (FDA) approved a new oral drug, the selective small molecule splenic tyrosine kinase inhibitor, fostamatinib, for medical use in patients with refractory ITP. And in 2021, a bioavailable small molecule thrombopoietin receptor agonist, avatrombopag, for the treatment of adult patients with chronic ITP who have had an insufficient response to previous therapy. Both drugs are not registered in Russia for the treatment of ITP. This is perspective.
If various treatment options are unsuccessful in subsequent lines of therapy, it is recommended to use a non-implementing method or carry out complex therapy using immunosuppressants.
As a rule, modern methods of therapy still make it possible to achieve remission of varying durations or states of clinical compensation. But clear prognostic criteria for the course of the disease, response to therapy and disease outcomes have not yet been developed - due to the nature and unpredictable course of the disease.
When starting treatment for chronic, recurrent ITP, it is necessary to remember that the choice of therapy should be aimed at stopping bleeding of any location, improving the patient’s quality of life, and not at normalizing the platelet count at any cost.
In clinical practice, it is important to remember that therapy should always be selected individually for a particular patient, taking into account his age, comorbidity, concomitant pathology, and also taking into account the patient’s preferences. But our practice often collides with the objective realities of life.
According to E.Yu. Krasilnikova (head of the project office “Rare (orphan) diseases” of the National Research Institute of Public Health named after N.A. Semashko), when preparing the Annual Bulletin on rare (orphan) diseases9, information was received from 76 regions of the Russian Federation in which, as of January 1, 2021, There were 3,860 patients with ITP (873 of them were children), 2,069 people needed drug therapy (468 of them were children), 1,606 patients (of which 446 were children) received drug therapy. That is, pathogenetic treatment was provided to 42% of patients included in the Federal Register of Persons Suffering from Rare Life-Threatening Diseases.8
In fact, more than half of patients do not receive the intended orphan treatment. The quality of medical care, which includes diagnosis, determination of treatment tactics, correction and control over these indicators, became possible thanks to the development of a patient routing scheme, which represents the patient’s path from diagnosis to provision of necessary medications, that is, from the attending physician to the inclusion of patients in federal register list.
Our experience with regional hematologists is that they are all knowledgeable and respectful of patient routing pathways for orphan drugs. In difficult situations, they turn to federal centers to obtain a decision from a medical commission on expensive drugs. Providing only 42% of patients with ITP in the regions with modern, expensive orphan drugs is mainly due to insufficient funding in a number of regions. And this also has its explanation. The number of patients requiring expensive orphan drugs is increasing due to improved diagnostics. The only way out is to include ITP in the federal program for financing high-cost nosologies.
Thus, idiopathic thrombocytopenic purpura (ITP) is a rare (orphan) chronic, relapsing disease that significantly worsens the health and quality of life of patients as assessed by physical, social functioning, and mental state. Bleeding causes fear, anxiety and depression in them due to the short-term effect of the therapy and side effects of drugs during long-term treatment with corticosteroids and immunosuppressants.
ITP cannot be completely cured, but it can be effectively controlled. Modern drugs (thrombopoietin receptor agonists) that have appeared in recent years, with adequate choice of dose and control of the course of the disease, can quickly stop hemorrhagic syndrome, achieve remission of varying durations or a state of clinical compensation, prevent the development of severe side effects of treatment, improve the prognosis of the disease, which, naturally, not only increases the life expectancy of patients with orphan diseases, but also its quality. Therefore, it is very important to include them in the therapy of all patients who need it. Today, this equal accessibility is possible only with the inclusion of ITP in the federal program for financing high-cost nosologies.
Literature
- “On the fundamentals of protecting the health of citizens in the Russian Federation” No. 323-FZ dated November 21, 2011. RG, federal issue No. 263 (5639) (dated November 23, 2011). https://rg.ru/2011/11/23/zdorovie-dok.html
- Decree of the Government of the Russian Federation No. 403 of April 26, 2012 “On the procedure for maintaining the Federal Register of persons suffering from life-threatening and chronic progressive rare (orphan) diseases leading to a reduction in the life expectancy of citizens or their disability, and its regional segment.” May 2, 2012. https://www.garant.ru/products/ipo/prime/doc/70068888/
- Chernikov M.V., Kulikov S.M., M.A. Rusinov M.A. et al. Multinosological register of diseases of the blood system. Composition, structure, results of trial operation // Hematology and transfusiology. T. 59, No. 1, 2014, p. 30. Melikyan A.L., Egorova E.K., Pustovaya E.I., Kolosheinova T.I., Volodicheva E.M., Kaporskaya T.S., Ilyasov R.K., Shelekhova T.V., Fedorova N.A., Zotova I.I., Sycheva T.M., Kontievsky I.N., Shestopalova I.A., Kurkina N.V., Syrtseva E.B., Tarasenko E.V. Interim results of an epidemiological study of idiopathic thrombocytopenic purpura in adults in the Russian Federation // Hematology and Transfusiology. 2021. T 64. No. 4. P. 436‒446.
- Melikyan A.L., Pustovaya E.I., Egorova E.K., Kalinina M.V., Kolosheinova T.I., Subortseva I.N., Gilyazitdinova E.A., Dvirnyk V.N. Differential diagnosis of thrombocytopenia // Oncohematology. 2017;12(1):78‒87. https://doi.org/10.17650/1818-8346-2017-12-1-78-87
- Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2021 guidelines for immune thrombocytopenia. 2019;3(23):3829‒3866. doi:10.1182/bloodadvances.201900096
- Melikyan A.L., Pustovaya E.I., Egorova E.K., Kalinina M.V., Kolosheinova T.I., Subortseva I.N., Gilyazitdinova E.A., Dvirnyk V.N. Differential diagnosis of thrombocytopenia // Oncohematology. 2017;12(1):78‒87. https://doi.org/10.17650/1818-8346-2017-12-1-78-87
- Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780‒3817. doi:10.1182/bloodadvances.2019000812
- Annual bulletin of the expert council on rare (orphan) diseases. https://komitet2-.km.duma.gov.ru/upload/site21/Byulleten_po_redkim_zabolevaniyam_2020.pdf
- Clinical guidelines “Idiopathic thrombocytopenic purpura (ITP) in adults” (approved by the Ministry of Health of Russia), 2021. https://npngo.ru/uploads/media_document/283/5eb37419-9276-4e9a-b075-0e26a788f623.pdf
Causes
Why this disease occurs is unknown. Doctors talk about hereditary predisposition and the negative effects of the following factors:
- bacterial and viral infections;
- vaccinations;
- excessive exposure to solar radiation (insolation);
- general hypothermia of the body;
- some medications (Indomethacin, Furosemide).
On the surface of platelets there are molecular complexes called antigens. If a foreign antigen enters the body, the immune system immediately begins to produce specific antibodies. By interacting with antigens, they destroy the cells on whose surface they are located.
In essential thrombocytopenia, the spleen produces antibodies to its platelet antigens. They are fixed on platelet membranes and, as it were, mark them. As they pass through the spleen, blood cells are captured and destroyed.
Due to a decrease in the number of platelets, the liver begins to produce them more. The rate of maturation of megakaryocytes and the formation of platelets from them in the red bone marrow increases significantly. But gradually the compensatory capabilities of the bone marrow become thinner, and the disease makes itself felt.
If autoimmune thrombocytopenia is diagnosed during pregnancy, this means that antibodies to one’s own platelets cross the placental barrier and destroy normal fetal platelets. As a result, the child may be born sick.
Thrombocytopenia consumption
Platelets are activated in the vascular bed, and the blood clotting mechanism is activated. Due to the increased consumption of platelets, the red bone marrow begins to actively produce them, and if the cause of the pathology is not eliminated in time, their level in the blood will decrease to a critical level.
The activation of formed elements is caused by:
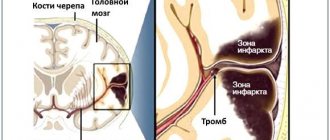
- Thrombotic thrombocytopenic purpura. Caused by an insufficient amount of prostacyclin in the blood. This anticoagulant factor is produced by the endothelium and prevents platelets from sticking to each other. When its release is disrupted, local platelet activation occurs and microthrombi form. Vessels are damaged and intravascular hemolysis develops.
- Disseminated intravascular coagulation syndrome. Appears as a result of severe damage to internal organs or tissues (infections, burns, injuries, surgeries, etc.), due to which the blood coagulation system is depleted. Numerous blood clots form in the vascular bed. They clog small vessels, preventing normal blood supply to the kidneys, brain and other organs. In response, the anticoagulant system is activated, aimed at restoring blood flow by breaking up blood clots. Over time, the blood completely loses its ability to clot. Severe internal bleeding may occur, which can be fatal.
- Hemolytic-uremic syndrome. Associated with intestinal infections, hereditary predisposition, systemic diseases and the use of certain medications. Bacterial toxins are released into the blood. They damage the vascular endothelium and activate platelets by attaching them to damaged areas. As a result, microthrombi are formed and microcirculation of internal organs is disrupted.
Thrombocytopenia redistribution
Normally, about 30% of all platelets are deposited in the spleen. In some diseases, the organ greatly increases in size, which is why up to 90% of the formed elements of blood are deposited in it. The disease can be caused by cirrhosis of the liver, systemic lupus erythematosus, infections, tumors, and alcoholism.
Thrombocytopenia dilution
Develops in patients who are transfused with large volumes of plasma, fluids, red blood cells, plasma substitutes, without replacing platelets. As a result, the concentration of the latter decreases to such a level that even their release from the depot is not able to maintain the normal functioning of the coagulation system.
Treatment of pathology
Treatment of thrombocytopenic purpura is aimed at:
- restoration of normal platelet levels and adequate bone marrow activity;
- prevention of bleeding;
- removal of autoimmune complexes from the bloodstream.
The treatment regimen is developed for each patient individually and depends on the severity of the patient’s condition and the degree of progression of TP.
Pharmacotherapy
Treatment with corticosteroids, thrombopoietin receptor agonists (stimulation of hematopoiesis), immunosuppressive drugs, and intravenous immunoglobulins is used.
Symptoms of thrombocytopenia
The mechanism for the development of thrombocytopenia symptoms is always the same. Due to a decrease in platelet concentration, the nutrition of the walls of blood vessels is disrupted, and capillary fragility increases. At a certain moment, due to a minor physical factor or spontaneously, the integrity of the capillaries is disrupted, and severe bleeding occurs (this is usually depicted in the photo as thrombocypenia). The lack of platelets prevents the formation of a platelet plug in the burst vessels, so blood flows from the circulation into the surrounding tissues.
The main symptoms of thrombocytopenia:
- Purpura (bleeding into mucous membranes, skin). Small red spots form on the body. They do not disappear when pressed, do not hurt and do not rise above the surface of the skin. Their size can range from a few millimeters to several centimeters in diameter. At the same time, bruises may appear.
- Bleeding gums. It forms on a large surface and does not stop for a long time.
- Frequent nosebleeds. Can be caused by sneezing, microtrauma, colds. The blood that comes out is bright red. Sometimes the bleeding cannot be stopped for an hour.
- Hematuria (blood in the urine). Occurs due to hemorrhage in the mucous membranes of the bladder or urinary tract.
- Gastric and intestinal bleeding. They are a consequence of increased fragility of the vessels of the gastrointestinal mucosa, traumatizing it with rough food. The stool becomes red and bloody vomiting may occur.
- Heavy and prolonged menstrual bleeding.
If you experience similar symptoms,
consult your doctor . It is easier to prevent a disease than to deal with the consequences.
Clinical picture of Werlhof's disease
The disease begins gradually or acutely with the appearance of hemorrhagic syndrome. The type of bleeding in thrombocytopenic purpura is petechial-spotted (bruised). According to clinical manifestations, two variants of thrombocytopenic purpura are distinguished: “dry” - the patient experiences only cutaneous hemorrhagic syndrome; “wet” - hemorrhages combined with bleeding. Pathognomonic symptoms of thrombocytopenic purpura are hemorrhages in the skin, mucous membranes and bleeding. The absence of these signs raises doubts about the correctness of the diagnosis.
- Cutaneous hemorrhagic syndrome occurs in 100% of patients.
The number of ecchymoses varies from single to multiple. The main characteristics of cutaneous hemorrhagic syndrome in thrombocytopenic purpura are as follows. Discrepancy between the severity of hemorrhage and the degree of traumatic exposure; their spontaneous appearance is possible (mainly at night). - Polymorphism of hemorrhagic rashes (from petechiae to large hemorrhages).
- Polychrome skin hemorrhages (color from purple to blue-greenish and yellow, depending on how long ago they appeared), which is associated with the gradual conversion of hemoglobin through intermediate stages of decomposition into bilirubin.
- Asymmetry (no favorite localization) of hemorrhagic elements.
- Painless.
There are no characteristic changes in internal organs with thrombocytopenic purpura. Body temperature is usually normal. Sometimes tachycardia is detected, during auscultation of the heart - systolic murmur at the apex and at Botkin's point, weakening of the first tone, caused by anemia. An enlarged spleen is uncharacteristic and rather excludes the diagnosis of thrombocytopenic purpura.
According to the course, acute (lasting up to 6 months) and chronic (lasting more than 6 months) forms of the disease are distinguished. During the initial examination, it is impossible to determine the nature of the course of the disease. Depending on the degree of manifestation of the hemorrhagic syndrome and blood parameters during the disease, three periods are distinguished: hemorrhagic crisis, clinical remission and clinical-hematological remission.
- Hemorrhagic crisis is characterized by severe bleeding syndrome and significant changes in laboratory parameters.
- During clinical remission, hemorrhagic syndrome disappears, bleeding time is reduced, secondary changes in the blood coagulation system are reduced, but thrombocytopenia persists, although it is less pronounced than during a hemorrhagic crisis.
- Clinical and hematological remission implies not only the absence of bleeding, but also the normalization of laboratory parameters.
Diagnostics
The presence of thrombocytopenia in children and adults is indicated by a decrease in the number of platelets in the blood. To make an accurate diagnosis and prescribe quality treatment, additional studies are carried out, which can be completed in any modern hematology clinic
:
- general blood analysis;
- Duke bleeding time assessment;
- puncture of red bone marrow (allows you to study the state of the hematopoietic system);
- determination of blood clotting time;
- genetic research (carried out if hereditary thrombocytopenia is suspected);
- determination of antibodies in the blood (clarification of the ratio of antibodies and platelets);
- Ultrasound (makes it possible to study the density and structure of internal organs, determine the size of the liver and spleen);
- MRI (to obtain layer-by-layer images of blood vessels and internal organs).
Diagnosis of the disease
Consultation with a hematologist
The doctor carefully examines and interviews the patient, finding out in detail the history of the disease, family history, etc.
Laboratory diagnostics
- general clinical blood test;
- biochemical blood test;
- determination of immunoglobulins;
- coagulogram;
- testing for infectious diseases (HIV, Helicobacter, herpes, etc.);
- determination of autoantibodies to platelets in peripheral blood;
- thrombophilia markers;
- testing for antiphospholipid syndrome;
- study of thyroid function.
Instrumental methods
- Ultrasound of the abdominal organs;
- Ultrasound of the thyroid gland;
- EchoCG.
All studies are carried out by experienced specialists using expert-class equipment, which ensures diagnostic accuracy.
Treatment of thrombocytopenia
Thrombocytopenia is treated by a hematologist. First, the patient’s condition is assessed and the severity of the hemorrhagic syndrome is studied.
- With mild severity (50,000-150,000 platelets per μl of blood), the capillary walls are in normal condition, so blood does not leave the vascular bed. Severe bleeding does not develop. Therefore, the problem can be solved without medications. The cause of the disease is being determined. The doctor adheres to a wait-and-see approach. The patient is not hospitalized.
- With moderate severity (25,000-50,000 platelets per μl of blood), hemorrhages in the oral mucosa and nosebleeds are observed. With minor bruises, large bruises form. If there is a risk of bleeding (presence of a stomach ulcer, playing professional sports, etc.), drug therapy is carried out. The patient can undergo treatment at home.
- With severe thrombocypenia (less than 20,000 platelets per μl of blood), spontaneous bleeding occurs in the mucous membranes of the mouth and skin. Hemorrhagic syndrome appears. Prescription of complex drug therapy is mandatory. The patient is hospitalized.
Treatment of thrombocytopenia with drugs
Drug treatment for any form of thrombocytopenia is aimed at:
- stopping bleeding;
- eliminating the cause;
- treatment of the disease that caused the problem.
Patients are usually prescribed medications:
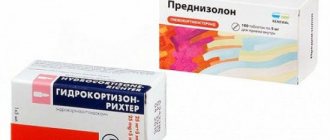
- "Prednisolone";
- "Intraglobulin", "Imbiogam";
- "Revolade";
- "Vincristine";
- "Etamzilat";
- "Depo-Provera";
- vitamin B12.
Non-drug treatments for thrombocytopenia
Treatment of thrombocypenia can be carried out using therapeutic and surgical measures. Most often, hematologists resort to:
- Transfusion therapy. The patient is transfused with donor blood (plasma or platelets).
- Removal of the spleen. The spleen is the main source of antibodies in immune thrombocytopenia and the main site of platelet destruction, so sometimes it is completely or partially removed. In most patients, after removal of the spleen for thrombocytopenia, the condition returns to normal and the clinical symptoms of the disease disappear.
- Bone marrow transplant. First, the patient is prescribed large doses of anticancer drugs that suppress the immune system. This is done in order to prevent the immune system from reacting to the introduction of donor bone marrow, as well as to destroy tumor cells if we are talking about a tumor of the hematopoietic system.
Treatment of thrombocytopenia during pregnancy
Thrombocytopenia in pregnant women requires proper treatment. Typically, expectant mothers are prescribed short courses of glucocorticosteroids (Dexamethasone, Prednisolone). In the third trimester, they accelerate the maturation of the fetal lungs, which makes early delivery possible.
If glucocorticosteroids are ineffective, immunoglobulin is administered intravenously. During the entire pregnancy it is used three to four times, and then again immediately after childbirth. In emergency situations, platelet transfusions are used.
If all the measures taken do not eliminate the symptoms of the disease, the spleen is removed in the second trimester (surgery is especially often resorted to if severe gestational thrombocytopenia is diagnosed in the first trimester). Ideally, surgery should be performed laparoscopically.
The issue of delivery is always decided on an individual basis. Thrombocytopenia during childbirth can lead to severe bleeding, so doctors carefully weigh the pros and cons. A cesarean section is considered less traumatic for the female body.
Treatment of thrombocytopenia with folk remedies
Thrombocytopenia is treated with the leaves of nettle, yarrow, and strawberry. Herbs are brewed with boiling water and drunk chilled, 1/4 cup 3-4 times a day.
Sesame oil has proven itself well for thrombocytopenia. It should be taken one tablespoon 3 times a day.
4.Diagnostics and treatment
Diagnosis of ITP
is based on thrombocytopenia, which can be easily detected by a clinical blood test or a coagulogram - a clotting test. It is more difficult (and more important) to differentiate this disease from other diseases with thrombocytopenic symptoms.
During the period of exacerbation of ITP, a positive reaction to a number of tests is observed:
- tourniquet test (positive if, when applying a tourniquet to the shoulder, small hemorrhages form on the skin of the shoulder and forearm),
- hammer test (positive if bruises occur when moderately tapping the skin with a hammer),
- pinch test (positive if pinching the skin under the collarbone within 24 hours produces petichiae or bruising),
- prick test (positive if, after an injection with a scarifier, the wound bleeds for more than 4 minutes).
Treatment of idiopathic thrombocytopenic purpura
most often symptomatic. If blood clotting rates are low, the patient is hospitalized. Bed rest is indicated. For anemia and prolonged bleeding, appropriate therapy is used. As the severity of the condition decreases, you are gradually allowed to get up and move.
Typically, the disease of idiopathic thrombocytopenic purpura sooner or later ends in spontaneous self-healing or long-term remission. Only in 1-2% of cases the disease leads to cerebral hemorrhage with a fatal outcome, and that is why secondary prevention includes reducing physical activity, gentle activity, and special monitoring during vaccinations, surgical treatment, and dental procedures. It is necessary to know the factors that reduce platelet production and avoid exposure to them.