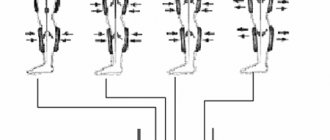
Symptoms of hypertrophic cardiomyopathy
Many people with HCM have either no or only mild symptoms and live normal lives. In other patients, symptoms may progress and worsen, thereby worsening heart function. Symptoms of hypertrophic cardiomyopathy can occur at any age and include:
- pain or pressure in the chest (usually occurs with physical exertion, but can also occur while resting or after eating)
- difficulty breathing (shortness of breath)
- fatigue (feeling extremely tired)
- fainting (due to irregular heart rhythms, abnormal functioning of blood vessels, or for no reason at all)
- rapid heartbeat (due to cardiac arrhythmias, usually ventricular extrasystoles, paroxysms of ventricular tachycardia, supraventricular arrhythmias are also common)
- sudden death (approximately 30% of patients have no complaints at all; sudden death may be the first and only manifestation of the disease.
Diagnostics
- An echocardiogram (ultrasound of the heart) is the most common test for diagnosing hypertrophic cardiomyopathy. With an echocardiogram, your doctor can see the thickness of your heart muscle, whether blood flow is abnormal, and the condition of your heart valves. An echocardiogram uses sound waves to produce an image of the heart. Echocardiography allows the doctor to see the heart in motion—as the ventricles contract and valves open and close.
Types of echocardiogram include:- Transthoracic echocardiography. This is a standard echocardiogram. The ultrasound doctor works using an ultrasound wave sensor, moving it along the surface of the chest. The sensor detects sound waves reflected from the heart. The computer converts them into moving images on the monitor.
Transesophageal echocardiography. In this type of echocardiography, a flexible tube containing a transducer is passed into the esophagus. From there, more detailed images of your heart can be obtained. Your doctor may order a transesophageal echocardiogram if it is difficult to get a clear picture of your heart during a standard ultrasound.
- Electrocardiogram (ECG). This test records the electrical activity of your heart. This is done to detect abnormal electrical signals that may result from thickening of the heart muscle.
- Holter monitoring. This is a simple test in which an ECG is recorded continuously for one or two days. It is used to detect abnormal heart rhythm.
- Coronary angiography. Sometimes, a doctor may use this test to rule out other heart pathologies. The catheter is inserted through a puncture into the artery in the thigh. Then you are carefully guided to the chambers of your heart under the guidance of an X-ray machine, which shows real-time images of your heart.
- MRI of the heart. Cardiac magnetic resonance imaging (MRI) is a method of producing images using magnetic fields and radio waves. Cardiac MRI is often used in addition to echocardiography, especially if echocardiographic findings are inconclusive.
- The question of genetic testing is determined by the doctor in each specific case.
If you have parents, siblings, or children with hypertrophic cardiomyopathy, experts recommend that you be regularly screened for signs of this condition.
The recommendation is to have an echocardiogram (ultrasound of the heart) and an electrocardiogram once a year. Over time, this diagnosis can be performed once every 5 years.
Recommendations
Diet
Drinking plenty of fluids is very important. Drink at least 6 to 8 glasses of water per day. In hot weather, you should increase your fluid intake. Fluids and salt restrictions are necessary for patients with heart failure. Talk to your healthcare provider about how specific liquids and diet drinks affect your body, including information about the effects of alcohol and caffeine.
Physical exercise
Your doctor will tell you whether exercise is recommended for you or not. Most people with cardiomyopathy are offered light aerobic exercise. However, your doctor may ask you to avoid any physical activity based on your symptoms and the severity of your illness. As with dilated cardiomyopathy, heavy lifting is not recommended
Regular doctor visits
Patients with HCM should visit a cardiologist at least once a year to monitor their condition. Visits to the doctor may be more frequent when hypertrophic cardiomyopathy is first diagnosed.
Causes of the disease
Hypertrophic cardiomyopathy is usually caused by gene mutations. These mutations are thought to cause the heart muscle to grow abnormally thick. People with hypertrophic cardiomyopathy also have altered alignment of the heart muscle fibers. And the cells in the fibers stop contracting synchronously, which often leads to heart rhythm disturbances.
The severity of hypertrophic cardiomyopathy varies widely. Most people with hypertrophic cardiomyopathy have a form of the disease in which the interventricular septum (the partition between the two lower chambers of the heart) becomes significantly enlarged, impeding blood flow. This condition is called hypertrophic obstructive cardiomyopathy.
Another common form of the disease is cardiomyopathy without significant obstruction to blood flow. However, in this case, the elasticity of the heart muscle suffers, the myocardium becomes rigid, which reduces the amount of blood ejected by the heart into the vascular system with each contraction. This form is called non-obstructive hypertrophic cardiomyopathy.
What drugs are used to treat hypertrophic cardiomyopathy?
Medicines used to treat symptoms and prevent further complications of HCM take the strain off the heart and reduce barriers to efficient blood circulation in the body. In the treatment of hypertrophic cardiomyopathy, medications based on beta-blockers are also used, which can reduce the load on the heart by lowering pressure and pulse, reducing myocardial hypertrophy. Beta blockers also have antiarrhythmic effects, including the ability to reduce the risk of fatal arrhythmias and sudden death.
Your doctor may ask you to avoid certain medications, such as nitrates (which lower blood pressure) or digoxin (which increases the force of your heart's contractions).
Your doctor will advise which medications are best for you.
Surgical procedures used to treat hypertrophic cardiomyopathy
Surgical techniques used to treat HCM include:
Transaortic septal myectomy. This technique involves reducing obstruction by reducing the thickness of the interventricular septum.
Transcatheter septal alcohol ablation. The introduction of sclerosing substances such as alcohol solutions, the creation of infarction zones in the interventricular septum.
Implantation of an artificial mitral heart valve. This type of surgery is offered to patients at high risk for life-threatening arrhythmias and sudden cardiac death. The mitral valve constantly monitors the heart rhythm. When it detects an abnormal, very fast heart rhythm, by supplying energy to the heart muscle, it causes the heart to beat at a normal rhythm.
Hypertrophic cardiomyopathy
Treatment of HCM, a genetically determined disease usually recognized at an advanced stage, is largely symptomatic and palliative. The main objectives of treatment measures are not only the prevention and correction of the main clinical manifestations of the disease with improving the quality of life of patients, but also a positive impact on the prognosis, prevention of cases of VS and progression of the disease. Patients with HCM are advised to avoid significant physical activity, which is accompanied by tachycardia, an even greater deterioration in LV diastolic filling and an increase in the intraventricular pressure gradient in the LV outflow tract.
When choosing a treatment program, the risk of sudden death of these patients is assessed. High risk factors for sudden death in HCM include young age (less than 14 years); the presence in patients of fainting and severe ventricular arrhythmias (spontaneous sustained ventricular tachycardia, ventricular fibrillation), episodes of unsustained ventricular tachycardia according to the results of 24-hour ECG monitoring; inappropriate increase in blood pressure during the stress test; pronounced (more than 3 cm) hypertrophy of the LV myocardium; indication of HCM and/or sudden death in family history. The likelihood of sudden death increases if the patient has atrial fibrillation (paroxysmal, permanent tachyform of atrial fibrillation), severe myocardial ischemia, or obstruction of the LV outflow tract.
Of great importance is the detection of mutations associated with a severe prognosis in patients with familial HCM. Establishing a high risk of sudden death determines the need for more active treatment tactics (clarification of drug therapy, use of pacemakers, defibrillators-cardioverters, and cardiac surgery). The most adequate treatment measure is the implantation of a defibrillator-cardioverter for the purpose of primary or secondary prevention of life-threatening arrhythmias and improving the prognosis.
Conservative treatment
The basis of drug therapy for HCM is drugs with a negative inotropic effect: b-blockers and calcium channel blockers (verapamil). To treat heart rhythm disturbances common in this disease, disopyramide and amiodarone are used.
β-blockers remain the most effective group of drugs used in the treatment of HCM. They have a good symptomatic effect in relation to the main clinical manifestations: shortness of breath and palpitations, pain, including angina, in at least half of patients with HCM, which is mainly due to the ability of these drugs to reduce myocardial oxygen demand.
Due to the negative inotropic effect and reduced activation of the sympathoadrenal system during physical and emotional stress, b-blockers prevent the emergence or increase of the intraventricular pressure gradient in patients with latent and labile obstruction, without significantly affecting the magnitude of this gradient at rest. The ability of b-blockers to improve the functional status of patients during a course and long-term use has been convincingly proven. Although the drugs do not directly affect myocardial diastolic relaxation, they may improve LV filling indirectly by reducing heart rate (HR) and preventing cardiac ischemia.
There is evidence in the literature confirming the ability of b-blockers to restrain and even reverse the development of myocardial hypertrophy. However, the symptomatic improvement caused by b-blockers is not accompanied by regression of LV hypertrophy and improvement in patient survival. Although the effect of these drugs in relieving and preventing ventricular and supraventricular arrhythmias and sudden death has not been proven, a number of experts still consider it advisable to prescribe them prophylactically to patients with high-risk HCM, including young patients with a family history of VS.
Preference is given to b-blockers without intrinsic sympathomimetic activity. The greatest experience has been accumulated in the use of propranolol (obzidan, anaprilin). It is prescribed starting at 20 mg 3–4 times a day, with a gradual increase in dose under the control of pulse and blood pressure (BP) to the maximum tolerated, in most cases up to 120–240 mg/day. You should strive to use the highest possible doses of the drug, because the lack of effect of b-blocker therapy is associated with insufficient dosage. Increasing dosages significantly increases the risk of known side effects.
Currently, the possibility of effective use of long-acting cardioselective b-blockers (atenolol, concor) is being widely studied. There is a point of view that cardioselective b-blockers in patients with HCM do not have advantages over non-selective ones, because in large doses selectivity is lost. Recommended for use in patients with HCM with severe supraventricular and ventricular arrhythmias, sotalol combines the properties of non-selective b-blockers and class III antiarrhythmic drugs (cordarone-like effect).
The use of slow calcium channel blockers in HCM is based on reducing the level of free calcium in cardiomyocytes and leveling the asynchrony of their contraction, improving myocardial relaxation and reducing its contractility, and suppressing the processes of myocardial hypertrophy. Due to the greatest severity of the negative inotropic effect and the most optimal profile of pharmacological properties, the drug of choice is verapamil (isoptin, finoptin).
It provides a symptomatic effect in 65–80% of patients, including cases of refractoriness to treatment with b-blockers, which is due to the ability of the drug to reduce myocardial ischemia, including painless, and improve its diastolic relaxation and LV compliance. This property of verapamil ensures an increase in patients' tolerance to physical activity and a decrease in the subaortic pressure gradient at rest, with a lower ability to reduce intraventricular obstruction during physical and emotional stress and provocation with isoproterenol compared to b-blockers. At the same time, verapamil reduces peripheral vascular resistance due to its vasodilator effect. However, severe complications of pharmacotherapy with verapamil develop in patients with non-obstructive HCM with high pressure in the left atrium, in whom they are caused by the negative inotropic effect of the drug. Hence, the importance of caution when starting treatment with verapamil in this category of patients is obvious.
Prescription of the drug should begin in a hospital setting with small doses - 20-40 mg 3 times a day with a gradual increase if well tolerated until the resting heart rate decreases to 50-60 per minute. The clinical effect usually occurs when taking at least 160–240 mg of the drug per day, and prolonged forms (isoptin retard, verogalide retard) are more convenient for long-term use. Taking into account the beneficial effect of verapamil on diastolic function and the magnitude of the subaortic pressure gradient in the LV, the proven ability to increase the survival of patients with HCM compared with placebo, its prophylactic administration in asymptomatic patients with high-risk HCM is advisable.
The place of diltiazem in the treatment of HCM has not been fully determined. There is evidence that at an average dose of 180 mg/day for 3 doses, it has the same pronounced beneficial effect on LV diastolic filling as 240 mg verapamil and the same symptomatic effect, but to a lesser extent improves the physical performance of patients [53]. It should be noted. that b-blockers (with the exception of sotalol) and calcium antagonists have weak antiarrhythmic activity, while the frequency of dangerous ventricular and supraventricular arrhythmias in patients with HCM is extremely high. Therefore, it is important to use antiarrhythmic drugs in this category of patients, among which the most popular and recommended by leading experts is disopyramide.
Disopyramide (rhythmilen), a class IA antiarrhythmic, has a pronounced negative inotropic effect; in patients with HCM it can reduce the level of obstruction of the LV outflow tract and have a positive effect on the structure of diastole. The effectiveness of long-term treatment with disopyramide has been proven in patients with HCM with moderate LV outflow obstruction. It is especially beneficial to use this drug in patients with ventricular arrhythmias. The initial dose is usually 400 mg per day with a gradual increase to 800 mg. In this case, it is necessary to monitor the duration of the QT interval using an ECG. An equally effective treatment and prevention of ventricular and supraventricular arrhythmias in HCM is amiodarone (cordarone), which, along with antiarrhythmic activity, somewhat reduces hypercontractility and myocardial ischemia. Its ability to prevent sudden death in patients with HCM has been demonstrated.
Treatment with amiodarone begins with saturating doses (600–1200 mg/day) for 3–7 days with a gradual reduction to maintenance (preferably 200 mg/day or less) as the heart rate decreases. Considering the deposition of the drug in tissues with possible dysfunction of the thyroid gland, the development of pneumofibrosis, damage to the cornea, skin and liver, with its long-term (more than 10-12 months) use, regular monitoring of the condition of these “vulnerable” organs is necessary in order to early identify possible complications of pharmacotherapy.
For HCM, combinations of drugs with a negative inotropic effect are possible: b-blockers and calcium antagonists, b-blockers and disopyramide. Signs of venous congestion in the lungs, including nocturnal attacks of cardiac asthma, are not uncommon in HCM and in most cases are due to LV diastolic dysfunction. Such patients are treated with b-blockers or calcium antagonists in combination with careful use of saluretics. Peripheral vasodilators and cardiac glycosides should be avoided due to the risk of worsening LV diastolic filling and a sharp decrease in cardiac output.
Atrial fibrillation and flutter are observed in 10–30% of patients with HCM and pose a risk of developing or worsening cardiohemodynamic disorders, the occurrence of thromboembolism, as well as an increased risk of developing ventricular fibrillation. As a result, in patients with HCM, paroxysmal supraventricular arrhythmias are classified as potentially fatal, and the rapid restoration of sinus rhythm and the prevention of repeated paroxysms becomes especially important. To relieve paroxysms of atrial fibrillation, antiarrhythmic drugs of group IA, amiodarone, b-blockers, verapamil and digoxin are used. If they are ineffective, electrical pulse therapy is performed.
For permanent atrial fibrillation, b-blockers or verapamil in combination with digoxin are used to control heart rate. This is the only case when patients with obstructive HCM can be prescribed cardiac glycosides without fear of increasing the intraventricular pressure gradient. Since atrial fibrillation in patients with HCM is associated with a high risk of systemic thromboembolism, after its development it is necessary to begin therapy with anticoagulants, which, with a permanent form of atrial fibrillation, are taken indefinitely.
In a significant number of patients with HCM, traditional pharmacotherapy does not effectively control the symptoms of the disease, and patients are not satisfied with the low quality of life. In such cases, it is necessary to decide on the possibility of using other, non-drug treatment approaches. In this case, further tactics are determined separately in patients with obstructive and non-obstructive forms of HCM. Contrary to popular belief, progressive systolic dysfunction and severe HF associated with LV remodeling often develop at an advanced stage of the pathological process (mainly in the non-obstructive form of HCM).
This evolution of the disease is observed in 2-5% of patients with HCM and characterizes the final (“dilated”) stage of a severe and accelerated process, independent of the patient’s age and how long ago the disease manifested itself. The increase in LV systolic size usually precedes and prevails over diastolic expansion. The clinical features of this stage are severe, often refractory congestive heart failure and an exceptionally poor prognosis. The treatment strategy for such patients is changing and is based on the general principles of treatment of congestive heart failure, and includes careful administration of ACE inhibitors, angiotensin II receptor blockers, diuretics, cardiac glycosides, b-blockers and spironolactone. These patients are potential candidates for heart transplantation.
Surgery
In the absence of clinical effect from adequate drug therapy in patients with NYHA class III–IV and severe asymmetric IVS hypertrophy, subaortic pressure gradient equal to 50 mm. Hg Art. and more, surgical treatment is indicated. The classic technique is transaortic septal myectomy, proposed by AG Morrow (1988). In young patients with a family history of HCM with severe clinical manifestations and indications of early sudden death in relatives, the indications should be expanded. In some cardiac surgery centers in Europe and the USA, it is also performed in cases of significant latent obstruction. In general, at least 5% of all patients with HCM are potential candidates for surgical treatment.
The operation provides a good symptomatic effect with complete elimination or significant reduction of the intraventricular pressure gradient in 95% of patients and a significant reduction in end-diastolic pressure in the LV of most patients. Surgical mortality has now decreased significantly and is about 1–2%, which is comparable to the annual mortality rate with medical therapy (2–5%). Although in most cases earlier studies failed to detect a significant effect of surgical treatment of HCM on prognosis, S. Seiler et al. (1989) showed an improvement in the 10-year survival rate of operated patients to 84% compared to 67% in the group treated with medication.
In some cases, if there are additional indications to reduce the severity of obstruction and mitral regurgitation, valvuloplasty or mitral valve replacement with a low-profile prosthesis is performed simultaneously. Subsequent long-term therapy with verapamil can improve long-term results of surgery, as it provides improvement in LV diastolic function, which is not achieved with surgical treatment.
Currently, techniques different from classical transaortic septal myectomy have been developed and are being successfully used. In particular, at the National Center for Agricultural Agricultural Sciences named after A.N. Bakulev under the leadership of Academician of the Russian Academy of Sciences and Russian Academy of Medical Sciences L.A. Boqueria developed an original technique for excision of the zone of hypertrophied IVS from the conical part of the pancreas. This method of surgical correction of obstructive HCM is highly effective and can be the method of choice in cases of simultaneous obstruction of the outflow tracts of both ventricles, as well as in cases of midventricular LV obstruction.
In recent years, there has been growing interest in studying the possibility of using sequential dual-chamber pacing with a shortened atrioventricular delay as an alternative to surgical treatment of patients with obstructive HCM. The resulting change in the sequence of propagation of the wave of excitation and contraction of the ventricles first covers the apex and then the IVS, leading to a decrease in the subaortic gradient due to a decrease in regional contractility of the IVS and expansion of the LV outflow tract.
It is important to select the smallest delay time for the application of the ventricular impulse after the atrial impulse, which ensures premature depolarization of the apex of the heart without leading to a deterioration in cardiohemodynamics - a decrease in cardiac output and blood pressure. To do this, in some cases it is necessary to resort to lengthening the time of spontaneous atrioventricular conduction using therapy with b-blockers or verapamil, and sometimes ablation of the atrioventricular node is used.
Although early uncontrolled observations were encouraging, more recent randomized trials have shown that the symptomatic effect and reduction in subaortic pressure gradient achieved with pacing are relatively small (about 25%) and there are no significant changes in physical performance. It was not possible to detect a significant effect of cardiac pacing on the incidence of sudden death. Therefore, until the role of cardiac pacing in the treatment of obstructive HCM is clarified, extended clinical use of this method is not recommended.
Another alternative treatment for refractory obstructive HCM is transcatheter alcohol septal ablation. The technique involves infusion of 1–3 ml of 95% alcohol through a balloon catheter into the perforating septal branch, resulting in an infarction of the hypertrophied part of the IVS, involving from 3 to 10% of the mass of the LV myocardium (up to 20% of the mass of the IVS). This leads to a significant reduction in the severity of outflow tract obstruction and mitral insufficiency, objective and subjective symptoms of the disease. Moreover, in 5–10% of cases there is a need to implant a permanent pacemaker due to the development of high-degree atrioventricular block.
Currently, the positive effect of transcatheter ablation on prognosis has not been proven, and operative mortality (1–2%) does not differ from that during septal myectomy, which is currently considered the “gold standard” for the treatment of patients with HCM with severe symptoms and obstruction of the LV outflow tract. resistant to pharmacotherapy.
In advanced cases (with the development of mitralization of the defect, a significant decrease in EF and the formation of FC IV CHF, accompanied by calcification of the aortic valve and the aortic wall), radical surgery becomes very risky. In these cases, it is better to limit ourselves to the application of a valve-containing shunt between the LV apex and the aorta, called apico-aortic.
This operation was first developed and performed by Denton Cooly in 1975. He performed it with artificial circulation, introducing a rigid tip through the apical incision into the LV cavity, which passes into a valve-containing Dacron shunt implanted into the descending aorta. During the operation, a second LV outlet was created and the gradient at the aortic outlet decreased or disappeared. The aggravating point in this technique was artificial blood circulation, which we excluded by developing our own method of operation (V.P. Polyakov, V.V. Goryachev, A.V. Polyakov, 1998).
To do this, we divided the shunt into 2 branches. One branch from the left-side lateral approach in the 6th intercostal space was anastomosed with the descending aorta above the diaphragm and clamped. The second branch ended with a soft Teflon ring sewn to it. Using this ring, the shunt was firmly fixed to the avascular portion of the LV apex using U-shaped sutures on spacers. Then, with a specially designed resector inserted into the lumen of this branch, a section of the LV wall (corresponding to the internal lumen of the branch) was excised and removed along with the resector. We calculated the length of the shunt visually. An anastomosis was performed between the branches with a disc valve prosthesis fixed in it.
After removing the clamps, the anastomosis began to function and immediately the pressure gradient in the outlet section sharply decreased or disappeared altogether. In the 90s of the 20th century, we operated on 22 patients with HCM with FC III-IV CHF using this method. Mortality during 10 years of observation did not exceed 9%. All patients received anticoagulant therapy, violations of which were associated with shunt thrombosis in two patients during the first 3-5 years.
Thus, treatment tactics for HCM are quite complex and require an individual analysis of the entire complex of clinical, anamnestic, hemodynamic parameters, results of gene diagnostics and stratification of the risk of sudden death, assessment of the characteristics of the course of the disease and the effectiveness of the treatment options used. In general, rational pharmacotherapy in combination with surgical treatment and electrocardiotherapy makes it possible to obtain a good clinical effect, prevent the occurrence of severe complications and improve the prognosis in a significant proportion of patients with hypertrophic cardiomyopathy.
Sudden death and endocarditis in hypertrophic cardiomyopathy
A small number of people with HCM are at increased risk of sudden cardiac death. People who are included in this risk group:
- relatives of those who died from sudden cardiac death
- young people with HCM with multiple episodes of loss of consciousness
- patients with high blood pressure and arrhythmia
- patients with severe symptoms and heart failure
If you have two or more risk factors for sudden cardiac death, your doctor will prescribe medications to prevent the arrhythmia. Most patients with HCM have a low risk of sudden cardiac death. Talk to your doctor about any problems you may face with hypertrophic cardiomyopathy.
Risk factors
Hypertrophic cardiomyopathy affects men and women equally.
The disease is transmitted, as a rule, by inheritance. There is a 50% chance that the children of those who suffer from hypertrophic cardiomyopathy will inherit a genetic mutation that subsequently leads to the development of the disease. Siblings of those with hypertrophic cardiomyopathy are also at risk. As a result, close relatives of those suffering from hypertrophic cardiomyopathy are strongly advised to consult a doctor to rule out the disease.
Overcoming difficulties
The diagnosis - hypertrophic cardiomyopathy - does not cause positive emotions. However, it should not be a cause of depression either. Cardiomyopathy is a curable disease, the method of treatment depends on its type and severity.
Because hypertrophic cardiomyopathy is inherited, it cannot be prevented. However, doctors and scientists are learning more about the genetic mutations that cause the disease. And it is important to identify this condition as early as possible so that treatment and prevention of complications can significantly help your health.