Near-death experience: people's memories
The most controversial and frequently discussed topic is the memories of people saved from clinical death. The images that people see in this state are often similar: bright light, tunnels, sensations of flying or falling, leaving the body, talking with deities or deceased relatives. Many believe that since patients' visions are similar, this can serve as evidence of the existence of an afterlife. But science has its own explanation for this phenomenon.
Organizing a funeral yourself - instructions
Where does the feeling of bliss come from?
Many people who have experienced cardiac arrest have reported an incredible lightness that envelops their entire body, a feeling of euphoria, peace, and happiness that they have never experienced to such an extent before. But in fact, the reason for their appearance is quite understandable - during emergency situations, the body releases a certain amount of endorphins in order to “block” the strongest feeling of stress or pain. Considering that possible death is, in general, the most dangerous of all possible situations for the human body, the happiness hormone is produced in maximum quantities. In Germany, a study led by Alexander Wutzler proved that during clinical death the amount of serotonin increases at least three times.
Causes of acquired heart disease
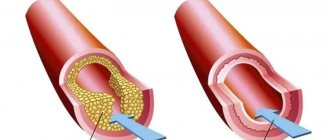
The most common causes of acquired heart disease are: rheumatism, sepsis, trauma, atherosclerosis, syphilis.
Types of congenital heart defects:
1) Heart defects that are caused by the presence of shunts:
- atrial septal defect;
- patent ductus arteriosus;
- ventricular septal defect.
2) Heart defects associated with the appearance of obstructions to blood flow and, as a result, an increase in the load on the heart:
- coarctation of the aorta;
- stenosis of the pulmonary or aortic valve.
A light in the end of a tunnel
A corridor or tunnel, a blinding light at the end - the most common visions of patients. All this is quite understandable if you have an idea about how our body works. The occipital lobe of the cerebral cortex is responsible for processing visual signals; its main functions are the analysis, perception and storage of visual information. And when a person dies (we already know that this does not happen with a snap of the fingers, but gradually), first we stop receiving the real picture, and only then the occipital lobe turns off. During this short period of time, during which the eyes no longer see, but the brain is still generating images, our field of vision narrows until only central (“tubular” vision) remains. The further oxygen starvation goes, the more rapidly our brain ceases to distinguish external signals from internal ones, thereby giving rise to a number of hallucinations that people see in a state of clinical death.
Why is heart disease dangerous?
A congenital heart defect may not be immediately apparent externally. The baby may appear healthy for some time after birth, and manifestations of the disease may occur until the third year of life. Manifestations of heart disease: a child’s delay in physical development, shortness of breath during physical exertion, pallor or even a blue tint to the skin.
Heart defects with a blue tint to the skin are characterized by attacks that occur suddenly. Restlessness, cyanosis appears, shortness of breath increases, and loss of consciousness is possible. This condition usually occurs in children under two years of age.
Defects with pallor are characterized by: developmental delays in the lower half of the body, headaches, dizziness, pain in the heart, legs and abdomen.
If you experience these symptoms, you need to make an appointment with a cardiologist who will conduct a number of necessary studies, give you an accurate diagnosis and help you cope with this illness.
Leaving the body
All organs suffer from severe hypoxia, including the vestibular analyzer. It is this organ that is responsible for the perception and analysis of information regarding the movement and position of our body in space. At the moment when our organs fail one after another, the vestibular apparatus also fails and is no longer able to adequately perceive information about the position of our body. On this basis, sensations of flight, “leaving” the body and ascending upward appear.
But for the record, many scientists generally do not consider out-of-body experiences to be unscientific. There is even unspoken statistics that say that 33% of all people at least once felt leaving their body and/or could see themselves from the outside. Especially often, such an experience is experienced by women giving birth: according to the results of studies conducted under the leadership of Dmitry Spivak (senior researcher of the neurophysiology of thinking and consciousness at the Human Brain Institute), about 9% of all examined women in labor consistently indicated a feeling of “leaving the body” and another 5.5 % on the appearance of these sensations after childbirth. Research has revealed that the appearance of these sensations is not affected in any way by blood loss during childbirth, painkillers, or the presence of mental illness. That is, out-of-body sensations are the result of extreme mental stress.
Diagnosis of sudden death of young people is one of the pressing problems in the work of a forensic medical expert. In the search line for the keywords “ sudden
death of young people
” in the Russian-language database eLibrary.ru, information about 13,259 sources is returned for the period from 2001 to 2021. In the English-language database Pubmed.com for a similar phrase “
sudden death of young people
” During the same time period, approximately 4,007 scientific articles were published. Thus, the number of annual publications indicates the relevance and unresolved nature of the identified problem.
According to the WHO definition, sudden cardiac death (SCD) includes cases of death occurring within 1 hour from the onset of signs of the disease that caused it in persons with known or unknown cardiac pathology [1, 2]. Some authors [1] indicate that the time interval from the onset of symptoms of the disease to death is 24 hours.
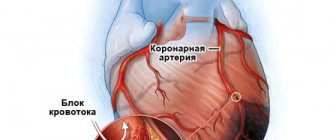
The main cause of SCD in 80% of cases is coronary heart disease (CHD) [1, 2]. In addition to coronary artery disease, SCD in young people is often caused by the presence of various types of cardiomyopathy [1–10]. According to the literature [11, 12], the likelihood of SCD occurring in people with structural heart pathology during the year is 7.5 times higher than in patients without such heart pathology.
The term " cardiomyopathy"
"(CM) was proposed in the middle of the 20th century and assumed that the patient had a heart pathology not associated with atherosclerosis of the coronary arteries. CMP is characterized by the presence of structural and functional disorders of the ventricular myocardium, which are not explained by limitation of blood flow due to coronary artery disease or high blood pressure [13, 14]. The first classifications of CMP as a heterogeneous pathology were based on objective data on the structure and function of the heart. Historically, this group of diseases has been divided into primary diseases, in which the heart is the only organ involved, and secondary forms, in which CMP reflects the development of manifestations of a systemic disease. The rapid development of genetics has expanded the understanding of the causes of CMP [15]. The concept has been put forward that CMP is caused by genetic disorders [13]. In 1996, WHO included heart diseases caused by cardiac dysfunction (heart rhythm disturbances) in the classification of cardiomyopathy [14]. However, the cause and pathogenesis of primary CMP are still not well understood.
For a forensic expert, the study of this pathology is of interest for the following reasons:
1. CMP often occurs hidden. The only clinical manifestation is acutely developed heart failure, leading to the death of the patient. Such a death is always suspected of being violent.
2. Not all forms of CMP have a clear morphological structure at both the macro- and microscopic levels. Consequently, the lack of a specific pathology structure (morphological markers) revealed at autopsy makes it difficult to establish the true cause of death and a search for new modern research methods is necessary.
One of the methods that is slowly being introduced into the practice of forensic experts is the method of molecular autopsy.
. This method, in particular, involves conducting a post-mortem genetic study to determine the carriage of mutant alleles in candidate genes for hereditary predisposition to sudden death associated with heart pathology in young people [16-19].
The purpose of the study was to analyze the literature on the interpretation of genetic studies in young people with CMP in combination with identified morphological features.
In economically developed countries, SCD ranges from 18.6 to 128 cases per 100,000 inhabitants per year [20]. Data from epidemiological studies show that IHD predominates in the structure of SCD. In people under 45 years of age, SCD is most often caused by the presence of various forms of CMP. C. Andersson and R. Vasan [21], when analyzing the death of persons under 35 years of age, most often and in equal percentages noted cases of sudden death caused by the presence of ischemic heart disease or various forms of cardiomyopathy. In 40% of postmortem diagnoses, sudden death of persons with CMP was associated with mutations in genes responsible for the structure of the heart, in 60% - with mutations in genes related to disturbances in electrolyte metabolism - the functioning of cardiomyocyte channels (so-called channelopathies) [22— 24]. It should be noted that cardiac muscle conduction disorders can be caused by mutations in genes responsible for protein synthesis in desmosomes [25–29].
A large number of classifications of ILC are known. Traditionally, types such as hypertrophic, dilated and arrhythmogenic CMP of the right ventricle of the heart, and restrictive CMP are distinguished [15].
Hypertrophic cardiomyopathy
(HCM), according to epidemiological studies, is the most common type of CMP. The incidence is 1 in 200–500 patients [14, 15], in the general population approximately 0.2% [30]. HCM is a monogenic disease associated with mutations in genes encoding the structure of components of the sarcomere apparatus [14, 31].
Morphologically, HCM is characterized by thickening of the wall of the left ventricle of the heart without expansion of its cavity. More than 20 years ago, it was recognized that HCM is inherited as an autosomal dominant trait. The most common mutations are in the following genes: MYH7
, encoding β-myosin heavy chains (30–50% of all hereditary HCM) [30],
MYBPC3 (
cardiac myosin binding protein C), troponin T, troponin I, myosin, myosin light chain, α-tropomyosin and cardiac α-actin. Analysis of the literature suggests that the relationship between genetic mutations in genes encoding sarcomere proteins, pathological phenotype and clinical outcomes remains poorly understood. To date, more than 1,400 mutations have been identified in 20 genes associated with HCM.
Dilated CMP
(DCM) is 0.57 per 100,000 patients per year, more often detected in children than in adolescents or young adults [15].
There are no reliable data on the incidence of DCM. About 40 genes associated with DCM have been identified [32], of which the most common mutations occur in genes encoding structural elements of the sarcomere ( MYH7, MYPN, MYBPC3, TNNT2
), cytoskeleton (
TTN
), sarcoplasmic reticulum (
PLN
), ion channels (
SCN5A
), nuclear membrane (
LMNA
), cell nucleus (
RBM20
) [33].
Thus, mutations in the TTN
cause increased oxidation in mitochondria, in candidate sarcomere genes they reduce sensitivity to calcium, and in
LMNA
and
TTN
lead to fibrosis [15].
A. Iuso et al. [34] noted that a mutation in the PPCS
(encoding phosphopantothenoyl cysteine synthase) causes autosomal recessively inherited DCM.
According to these authors, more genetic data are needed to identify the most important genetic mutations associated with DCM. P. Gourzi et al. [35] noted the association of DCM with a mutation in the LAMP2
, responsible for the expression of lysosomal membrane protein-2. Until recently, mutations in this gene were associated with hypertrophic cardiomyopathy in the context of Danon disease, characterized by weakening of the heart muscle (CHM), skeletal muscle weakness (myopathy) and intellectual impairment.
Arrhythmogenic right ventricular cardiomyopathy
( ARVCC )
occurs with a frequency of 1 case per 2000–5000 patients annually, more often in adolescents and young adults [15].
It is characterized by autosomal dominant inheritance [36]. There are 15 known genes whose mutations cause the development of ACMPC. Typically, ACMPC is inherited as an autosomal dominant disease, but cases of recessive inheritance have also been described [31, 36]. The use of NGS (next generation sequencing) methods has led to the identification of new mutagenic variants for ACMPC. The role of filamin C in the development of ACMPC indicates the genetic unity of ACMPC and other types of CMP. Putative pathogenic variants were found in the cadherin 2 gene, a protein involved in cell adhesion. A study of a cohort of patients with ACMPC allowed us to determine the key role of large genomic rearrangements in desmosomal genes [37]. Morphologically, patients with ACMPV have a heart of normal size. The name “ arrhythmogenic right ventricular cardiomyopathy ” is explained
by the initial assumption that pathomorphological changes are localized primarily in the right ventricle of the heart.
Histologically, ACMPC is characterized by the replacement of muscle tissue with fibrous tissue. Among the morphological features, it is interesting to note that genes associated with ACMPC are also associated with the disease palmoplantar keratoderma
and
the woolly hair disease
described by Naxos (cited in [37]).
Genetic linkage analysis and identification of homozygosity for a double-nucleotide deletion ( 2 1 5 7 del2
) in plakoglobin (
JUP
) identified inheritance of the syndrome as an autosomal recessive form with full penetration in adolescence, rather than an autosomal dominant one, as the main genetic cause [31].
This fact is not surprising, since among members of families with Naxos disease, heterozygous for the mutation in the same Pk2 1 5 7 del2
, clinical phenotypes are not detected.
It has been established that mutations in the coding of other genes of the main components of the desmosome cause ACMPC, including desmoplakin ( DSP
), plakophilin-2 (
PKP2
), desmoglin-2 (
DSG2
) and desmocollin-2 (
DSC2
) [31–42].
Desmosomes are complex structures consisting of proteins responsible for cell adhesion and the propagation of signals in cells. Thus, abnormalities in the structure of desmosomes are considered key in the pathogenesis of ACMPC and the classic form of the disease, which is RVCM, which was considered a disease of the desmosome
[37].
It is worth noting that ACMPP is caused not only by mutations in genes encoding desmosomal proteins. Mutations in the so-called non-desmosomal genes, which are also associated with ACMPC, were identified. Such mutations include changes in growth factor b3, transmembrane protein 43 ( TMEM4 3
), α-T-catenin (
CTNNA3
), titin (
TTN
), lamin A/C (
LAMINA-C
), phospholamban (
PLN
) and desmin (
DES
) . These findings for genes previously associated with other forms of CMP suggested a link between ACMPV and DCM at both the clinical and genetic levels [37].
One of the variants of the pathogenesis of ACMPV is associated with the proliferation of connective tissue in the wall of the right ventricle. When studying tissue markers of fibrosis using cardiovascular magnetic resonance imaging (imaging), late gadolinium enhancement was detected, regarded as a sign of an unfavorable outcome of the disease [31].
Restrictive CMP
occurs in less than 5% of all diagnosed CMP cases in European countries [15].
From the perspective of molecular biology, restrictive and hypertrophic CMP are associated with mutations in genes responsible for the structure of the sarcomere, while dilated CMP can be caused by mutations in genes regulating ion exchange and cytoskeletal structure [14].
Disorders associated with ion exchange in cardiomyocytes are called “channelopathies” [24, 43–46]. Channelopathies
usually observed in persons under 30 years of age. Clinical entities associated with channelopathies include long QT syndrome (LQTS), Brugada syndrome (BS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and short QT syndrome (SQTS) [47].
Most common LQTS
are Romano-Ward and Jervell-Lange-Nielsen syndromes, which are clinically combined with deafness.
It should be noted that LQTS is genetically heterogeneous, with at least eight genetic variants known. The most common is the LQTS1 subtype, which is caused by mutations in the KCNQ 1
, which encodes a subunit of the K-dependent channel responsible for the slow K+ current.
Mutations in the KCNH 2
, responsible for rapid K+ current, are the cause of LQTS2.
Heart rhythm disturbances occur during sympathetic activation of cardiac activity. This is precisely what explains the onset of SCD when performing physical activity in LQTS1. In LQTS2, SCD is associated with acute auditory stimuli [48]. LQTS in 10% of cases may be due to impaired Na+ metabolism. A mutation in the SCN 5 A
(LQTS3) gene, encoding the α-subunit of the sodium channel, promotes rapid clearance of Na+ during depolarization.
It was noted that a similar phenotype was observed for mutations in other genes ( CAV 3
,
SCN 4 B
and
SNTA 1
), directly or indirectly affecting sodium channel function.
Mutations in the KCNQ 1
,
KCNH 2
and
SCN 5 A
, causing LQTS1, 2 and 3, respectively, are the so-called core LQTS genes, and mutations in them suggest a high probability of congenital LQTS and are important for risk stratification [24]. It should be noted that there are differences in the frequency of occurrence of mutant variants depending on the population [49].
Patients with
B rugada
syndrome (SB) tend to develop fatal arrhythmias mainly during sleep, in the absence of myocardial ischemia, disturbances in electrolyte metabolism, and other structural changes in the heart.
ECG markers for SB are well known. The family history of SCD in SB is quite typical. The incidence of SB in European and American populations is 1 per 10,000 population. About 350 different variants of gene mutations associated with SB have been identified [50, 51]. SB is most often associated with mutations in the SCN5 A
. According to many authors [24, 52, 53], in 65% of cases the genetic substrate for BS is not identified.
Catecholaminergic polymorphic
ventricular tachycardia ( of Ca2
+ homeostasis.
This type of CMP is associated with mutations in the RyR2
, which encodes ryanodine receptors responsible for the release of calcium from the sarcoplasmic reticulum of cardiomyocytes.
The mutation is inherited in an autosomal dominant manner. CASQ2
gene, which is responsible for the synthesis of the protein calsequestrin, which performs the function of binding Ca2+ in the sarcoplasmic reticulum, are inherited in an autosomal recessive manner .
Another mutation associated with CPVT is in the TRDN
, which encodes triadin (a protein that binds to calsequestrin and RyR2 receptors). In the presence of mutations in these genes, SCD often occurs during emotional stress [54] or physical exertion [24].
The analysis of the scientific literature suggests that SCD is one of the main causes of mortality throughout the world. It is associated mainly with ischemic heart disease in older people and various forms of cardiomyopathy in young people. The peculiarity of the course of the disease in those suffering from various forms of cardiomyopathy is explained by the fact that acutely developed heart failure is often the first and only symptom of the existing disease. Sectional studies of deceased young people, as well as the statistical data obtained, indicate that in 1/3 of cases, during autopsy or additional histological studies, specific morphological markers of this pathology are not found. Currently, there are no accurate epidemiological data on the frequency of occurrence of various types of CMP in different populations due to the lack of uniform strict criteria for diagnosing this pathology [46]. It is possible that ICD-10 introduces uncertainty into the results of epidemiological studies. Probably, the epidemiology of this nosology should be structured in more detail. In particular, ICD-10 does not have sections devoted, for example, to arrhythmogenic right ventricular cardiomyopathy, which implies the use of the section “Other cardiomyopathies (I 42.8)” and “Cardiomyopathy, unspecified (I 42.9)”. In the last decade, there has been an understanding that primary CMPs are based on mutations in genes responsible for the synthesis of proteins that provide the structure of the sarcomere, desmosomes, or membrane channels that provide ion exchange in the cardiomyocyte. Genetic typing methods, namely the transition to the use of NGS typing, have made it possible to expand our understanding of the nature of CMP, especially in cases where the morphological substrate has not been determined [17]].
The authors of the article have no doubt that molecular genetic typing carried out after death for the carriage of mutations in genes associated with SCD has the right to be introduced into the practical work of a forensic expert.
It should be noted that with a large number of identified mutations, the problem of interpreting the results of genetic typing in the practice of a forensic expert involuntarily arises. Some authors [55] objectively express pessimism in the expert assessment of the obtained genetic results. In our opinion, one of the ways to objectively interpret the obtained genetic data is a thorough comprehensive analysis of the circumstances of the death of a young patient (victim), the collected family history, autopsy data and laboratory test results.
The authors declare
no conflict of interest.
1e-mail; https://orcid.org/0000-0001-5370-4931