Околосмертный опыт: воспоминания людей
Самая спорная и часто обсуждаемая тема – это воспоминания людей, спасённых от клинической смерти. Образы, которые видят люди в этом состоянии, зачастую схожи: яркий свет, тоннели, ощущения полёта или падения, выход из тела, разговор с божествами или умершими родными. Многие считают, что раз видения у пациентов схожи, это может служить доказательством существования загробной жизни. Но в науке есть своё объяснение этому явлению.
Самостоятельная организация похорон — инструкция
Откуда возникает ощущение блаженства
Многие люди, пережившие остановку сердца, рассказывали о невероятной лёгкости, окутывающей всё тело, ощущении эйфории, покоя, счастья, которых никогда прежде в такой степени не испытывали. Но на самом деле причина их появления вполне объяснима – во время экстренных ситуаций организм выделяет определённое количество эндорфинов, для того чтобы «заблокировать» сильнейшее ощущение стресса или боли. Учитывая то, что возможная смерть является для человеческого организма в общем-то самой опасной из всех возможных ситуаций, гормон счастья вырабатывается в максимальном количестве. В Германии в ходе исследования во главе с Александром Вутцлером было доказано, что во время клинической смерти количество серотонина увеличивается минимум в три раза.
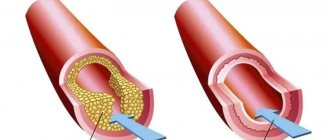
Причины приобретенного порока сердца
Самыми распространенными причинами приобретенного порока сердца являются: ревматизм, сепсис, травма, атеросклероз, сифилис.
Виды врожденных пороков сердца:
1) Пороки сердца, которые вызваны наличием шунтов:
- дефект межпредсердной перегородки;
- незаращение артериального протока;
- дефект межжелудочковой перегородки.
2) Пороки сердца, связанные с появлением препятствий кровотоку и, как следствие, увеличением нагрузки на сердце:
- коарктация аорты;
- стеноз легочного или аортального клапана.
Свет в конце тоннеля
Коридор или тоннель, ослепительный свет в конце – наиболее распространённые видения пациентов. Всё это вполне объяснимо, если иметь представления о работе нашего организма. За обработку зрительных сигналов отвечает затылочная доля коры головного мозга, её главные функции – это анализ, восприятие и хранение зрительной информации. И когда человек умирает (мы уже знаем, что происходит это не по щелчку пальцев, а всё же постепенно) сперва мы перестаём получать реальную картинку, а уже потом отключается затылочная доля. За этот короткий промежуток времени, в который глаза уже не видят, но мозг картинку всё ещё генерирует, наше поле зрения сужается до тех пор, пока не останется только центральное (“трубчатое” зрение). Чем дальше заходит кислородное голодание, тем стремительнее наш мозг перестаёт отличать внешние сигналы от внутренних, тем самым порождая ряд галлюцинаций, которые и видят люди в состоянии клинической смерти.
Чем опасен порок сердца?
Врожденный порок сердца может не сразу проявиться внешне. Некоторое время после рождения ребенок может выглядеть здоровым, и проявления заболевания могут происходить до третьего года жизни. Проявления порока сердца: отставания ребенка в физическом развитии, одышка при физических нагрузках, бледность или даже синий оттенок кожи.
Для пороков сердца с проявлением синего оттенка кожи характерны приступы, возникающие внезапно. Появляется беспокойство, цианоз, нарастает одышка, возможна потеря сознания. Такое состояние, как правило, встречается у детей до двух лет.
Для пороков с проявлением бледности характерны: отставание в развитии для нижней половины туловища, головные боли, головокружение, боль в сердце, ногах и животе.
Если вы ощущаете данные симптомы, необходимо записаться на консультацию к врачу-кардиологу, который проведет ряд необходимых исследований, поставит Вам точный диагноз и поможет вам справиться с эти недугом.
Выход из тела
От сильнейшей гипоксии страдают все органы, в том числе и вестибулярный анализатор. Именно этот орган отвечает за восприятие и анализ информации касательно движения и положения нашего тела в пространстве. В момент, когда наши органы один за другим дают сбои, вестибулярный аппарат тоже выходит из строя и уже не способен адекватно воспринимать информацию о положении нашего тела. На этой почве и появляются ощущения полёта, «выхода» из тела и вознесения ввысь.
Но к общему сведению, многие учёные в общем-то не считают внетелесные переживания антинаучными. Существует даже негласная статистика, в которой говорится, что 33% всех людей хотя бы единожды ощущали выход из своего тела и/или могли видеть себя со стороны. Особенно часто подобный опыт испытывают рожающие женщины: по результатам исследований, проводившимся под руководством Дмитрия Спивака (старший научный сотрудник нейрофизиологии мышления и сознания Института мозга человека), около 9% всех обследованных рожениц стабильно указывали на ощущение «выхода из тела» и ещё 5,5% на появление этих ощущений уже после родов. В ходе исследований выявили, что на появление этих ощущений никак не влияют кровопотери в родах, обезболивающие средства и наличие психических заболеваний. То есть внетелесные ощущения — результат предельного психического напряжения.
Диагностика внезапной смерти лиц молодого возраста является одной из актуальных проблем в работе судебно-медицинского эксперта. В поисковой строке по ключевым словам «внезапная
смертьмолодых
» в русскоязычной базе данных eLibrary.ru выдаются сведения о 13 259 источниках за период с 2001 по 2021 г. В англоязычной базе данных Pubmed.com по аналогичному словосочетанию «
sudden death of young
» за тот же временной промежуток опубликовано примерно 4007 научных статей. Таким образом, количество ежегодных публикаций свидетельствует об актуальности и нерешенности обозначенной проблемы.
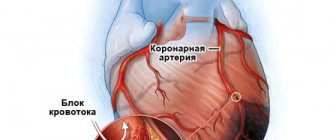
По определению ВОЗ, к внезапной сердечной смерти (ВСС) относятся случаи наступления летального исхода в течение 1 ч от появления признаков заболевания, ставшего его причиной, у лиц с известной или неизвестной сердечной патологией [1, 2]. Некоторые авторы [1] указывают, что временной промежуток от начала симптомов болезни до наступления смерти равен 24 ч.
Основной причиной ВСС в 80% случаев является ишемическая болезнь сердца (ИБС) [1, 2]. Кроме ИБС, у лиц молодого возраста ВСС часто обусловлена наличием различных видов кардиомиопатии [1—10]. По данным литературы [11, 12], вероятность наступления ВСС у лиц со структурной патологией сердца в течение года в 7,5 раза выше, чем у пациентов без такой патологии сердца.
Термин «кардиомиопатия
» (КМП) предложен в середине XX века и предполагал наличие у пациента патологии сердца, не связанной с атеросклерозом коронарных артерий. КМП характеризуется наличием структурных и функциональных нарушений миокарда желудочков, которые не объясняются ограничением кровотока вследствие ИБС или повышенного кровяного давления [13, 14]. Первые классификации КМП как гетерогенной патологии базировались на объективных данных структуры и функции сердца. Исторически эта группа заболеваний подразделялась на первичные заболевания, при которых сердце является единственным вовлеченным органом, и вторичные формы, при которых КМП отражает развитие проявлений системного заболевания. Бурное развитие генетики расширило понимание о причинах КМП [15]. Была выдвинута концепция, что КМП обусловлена генетическими нарушениями [13]. В 1996 г. ВОЗ включила в классификацию КМП болезни сердца, обусловленные сердечной дисфункцией (нарушение сердечного ритма) [14]. Тем не менее причина и патогенез первичной КМП до сих пор изучены недостаточно.
Для судебно-медицинского эксперта изучение данной патологии представляет интерес по следующим позициям:
1. Часто КМП протекает скрыто. Единственным клиническим проявлением является остро развившаяся сердечная недостаточность, приведшая к смерти пациента. Такая смерть всегда подозрительна на насильственную.
2. Не все формы КМП имеют четкую морфологическую структуру как на макро-, так и на микроскопическом уровне. Следовательно, отсутствие специфической структуры патологии (морфологические маркеры), выявляемой на вскрытии, затрудняет установление истинной причины смерти и необходим поиск новых современных методов исследования.
Один из методов, который медленно внедряется в практику судебно-медицинского эксперта, — метод молекулярной аутопсии
. Этот метод, в частности, предполагает проведение посмертного генетического исследования на предмет носительства мутантных аллелей в генах-кандидатах наследственной предрасположенности к внезапной смерти, связанной с патологией сердца у лиц молодого возраста [16—19].
Цель исследования — анализ литературы, посвященной вопросам интерпретации генетических исследований у лиц с КМП молодого возраста в сочетании с выявленными морфологическими особенностями.
В экономически развитых странах ВСС составляет от 18,6 до 128 случаев на 100 000 жителей в год [20]. Данные эпидемиологических исследований показывают, что в структуре ВСС преобладает ИБС. У лиц моложе 45 лет наиболее часто ВСС обусловлена наличием различных форм КМП. C. Andersson и R. Vasan [21] при анализе смерти лиц в возрасте до 35 лет наиболее часто и в равных процентных соотношениях отметили случаи внезапной смерти, обусловленной наличием ИБС или различных форм КМП. В 40% посмертно установленных диагнозов внезапная смерть лиц с КМП была связана с мутациями в генах, ответственных за структуру сердца, в 60% — с мутациями в генах, имеющих отношение к нарушению обмена электролитов — функционированию каналов кардиомиоцитов (так называемые каналопатии) [22—24]. Следует отметить, что нарушение проводимости сердечной мышцы может быть обусловлено мутациями в генах, ответственных за синтез белков в десмосомах [25—29].
Известно большое число классификаций КМП. Традиционно выделяют такие виды, как гипертрофическая, дилатационная и аритмогенная КМП правого желудочка сердца, рестриктивная КМП [15].
Гипертрофическая КМП
(ГКМП), по данным эпидемиологических исследований, — наиболее распространенный вид КМП. Частота встречаемости 1 на 200—500 пациентов [14, 15], в общей популяции примерно 0,2% [30]. ГКМП представляет собой моногенную болезнь, связанную с мутациями в генах, кодирующих строение компонентов аппарата саркомера [14, 31].
Морфологически ГКМП характеризуется утолщением стенки левого желудочка сердца без расширения его полости. Более 20 лет назад было признано, что ГКМП наследуются в виде аутосомно-доминантного признака. Наиболее часто встречаются мутации в следующих генах: MYH7
, кодирующем тяжелые цепи β-миозина (30—50% всех наследственных ГКМП) [30],
MYBPC3 (
миозинсвязывающий белок сердца C), тропонине T, тропонине I, миозине, легкой цепи миозина, α-тропомиозине и α-актине сердца. Анализ литературы свидетельствует, что связи между генетической мутацией в генах, кодирующих белки саркомера, патологическим фенотипом и клиническими исходами остаются плохо изученными. На сегодняшний день установлено более 1400 мутаций в 20 генах, ассоциированных с ГКМП.
Дилатационная КМП
(ДКМП) составляет 0,57 на 100 000 пациентов в год, чаще выявляется у детей, чем у подростков или лиц молодого возраста [15]. Надежных данных о частоте встречаемости ДКМП нет. Установлено около 40 генов, связанных с ДКМП [32], из них наиболее часто мутации встречаются в генах, кодирующих структурные элементы саркомера (
MYH7, MYPN, MYBPC3, TNNT2
), цитоскелета (
TTN
), саркоплазматической сети (
PLN
), ионных каналов (
SCN5A
), ядерной мембраны (
LMNA
), клеточного ядра (
RBM20
) [33]. Так, мутации в гене
TTN
вызывают повышенное окисление в митохондриях, в генах-кандидатах саркомера снижают чувствительность к кальцию, в
LMNA
и
TTN
приводят к фиброзу [15]. A. Iuso и соавт. [34] обратили внимание, что мутация в гене
PPCS
(кодирует фосфопантотеноил-цистеинсинтазу) вызывает аутосомно-рецессивно наследуемую ДКМП. По мнению этих авторов, необходимо больше генетических данных для выявления наиболее важных генетических мутаций, связанных с ДКМП. P. Gourzi и соавт. [35] отметили связь ДКМП с мутацией в гене
LAMP2
, ответственном за экспрессию лизосомального мембранного белка-2. До недавнего времени мутацию в данном гене связывали с гипертрофической КМП в контексте болезни Данона, характеризующейся ослаблением мышцы сердца (КМП), скелетных мышц (миопатия) и расстройством интеллекта.
Аритмогенная КМП правого
желудочка
(АКМППЖ) встречается с частотой 1 случай на 2000—5000 пациентов ежегодно, чаще у подростков и лиц молодого возраста [15]. Характеризуется аутосомно-доминантным наследованием [36]. Известно 15 генов, мутации в которых вызывают развитие АКМППЖ. Обычно АКМППЖ наследуется как аутосомно-доминантное заболевание, однако описаны случаи и рецессивного наследования [31, 36]. Использование методов NGS (секвенирование нового поколения) привело к идентификации новых мутагенных вариантов для АКМППЖ. Роль филамина C в развитии АКМППЖ свидетельствует о генетическом единстве АКМППЖ и других видов КМП. Предполагаемые патогенные варианты обнаружили в гене кадгерина 2 — белка, участвующего в клеточной адгезии. Исследование когорты пациентов с АКМППЖ позволило определить ключевую роль больших геномных перестроек в генах десмосом [37]. Морфологически у пациентов с АКМППЖ сердце обычных размеров. Название «
аритмогенная кардиомиопатия правогожелудочка
» объясняется первоначальным предположением, что патоморфологические изменения локализуются преимущественно в правом желудочке сердца. Гистологически АКМППЖ характеризуется замещением мышечной ткани фиброзной. Из морфологических признаков интересно отметить, что гены, связанные с АКМППЖ, также ассоциируются с заболеванием
palmoplantar keratoderma
и
болезнью шерстяных волос
, описанной Наксосом (цит. по [37]). Анализ генетической связи и идентификация гомозиготности для двунуклеотидной делеции (
2157del2
) в плакоглобине (
JUP
) позволил определить в качестве основной генетической причины наследование синдрома аутосомно-рецессивной формы с полной пенетрацией в подростковом возрасте, а не аутосомно-доминантной [31]. Этот факт не удивляет, так как среди членов семей с болезнью Наксоса, гетерозиготных по мутации в том же гене
Pk2157del2
, не выявляются клинические фенотипы.
Установлено, что мутации в кодировании других генов основных компонентов десмосомы вызывает АКМППЖ, включая десмоплакин (DSP
), плакофилин-2 (
PKP2
), десмоглин-2 (
DSG2
) и десмоколлин-2 (
DSC2
) [31—42]. Десмосомы представляют собой сложные структуры, состоящие из белков, ответственных за клеточную адгезию и распространение сигналов в клетках. Таким образом, аномалии в структуре десмосом рассматриваются в качестве ключевых в патогенезе АКМППЖ и классической формы заболевания, представляющей КМП правого желудочка, которая считалась
болезнью десмосомы
[37]. Стоит отметить, что АКМППЖ обусловлена не только мутациями в генах, кодирующих белки десмосом. Выявили мутации в так называемых недесмосомальных генах, ассоциируемые также с АКМППЖ. К таким мутациям относятся изменение фактора роста b3, трансмембранного белка 43 (
TMEM43
), α-Т-катенина (
CTNNA3
), титина (
TTN
), ламина A/C (
LAMINA-C
), фосфоламбана (
PLN
) и десмина (
DES
). Такие выводы для генов, ранее связанных с другими формами КМП, позволили говорить о связи между АКМППЖ и ДКМП как на клиническом, так и на генетическом уровне [37].
Один из вариантов патогенеза АКМППЖ связывают с разрастанием соединительной ткани в стенке правого желудочка. При изучении тканевых маркеров фиброза с помощью сердечно-сосудистого магнитно-резонансного исследования (томография) выявили позднее гадолиниевое усиление, расцениваемое как признак неблагоприятного исхода заболевания [31].
Рестриктивная КМП
встречается менее чем в 5% случаев от всех установленных КМП в европейских странах [15].
С позиции молекулярной биологии рестриктивная и гипертрофическая КМП связаны с мутациями в генах, ответственных за структуру саркомера, в то время как дилатационная КМП может быть обусловлена мутациями в генах, регулирующих обмен ионов и структуру цитоскелета [14].
Нарушения, связанные с обменом ионов в кардиомиоцитах, получили название «каналопатии» [24, 43—46]. Каналопатии
наблюдаются обычно у лиц в возрасте до 30 лет. К клиническим формам, которые связывают с каналопатиями, относят длинный QT-синдром (LQTS), синдром Бругада (СБ), катехоламинергическую полиморфную желудочковую тахикардию (КПЖТ) и короткий QT-синдром (SQTS) [47].
Наиболее распространенные варианты LQTS
— это синдромы Романо—Варда и Джервелла—Ланге—Нильсена, которые клинически сочетаются с глухотой. Следует отметить, что LQTS генетически неоднороден, известно не менее восьми генетических вариантов. Наиболее распространенный — подтип LQTS1, который обусловлен мутациями в гене
KCNQ1
, кодирующем субъединицу K-зависимого канала, ответственного за медленный ток K+. Мутации в гене
KCNH2
, ответственном за быстрый ток К+, являются причиной LQTS2. Нарушение сердечного ритма отмечается во время симпатической активации сердечной деятельности. Именно этим объясняется наступление ВСС при выполнении физических нагрузок при LQTS1. При LQTS2 ВСС ассоциируется с резкими звуковыми раздражителями [48]. LQTS в 10% случаев может быть обусловлен нарушением обмена Na+. Мутация в гене
SCN5A
(LQTS3), кодирующем α-субъединицу натриевого канала, способствует быстрому выведению Na+ при деполяризации. Отмечено, что похожий фенотип наблюдался для мутаций в других генах (
CAV3
,
SCN4B
и
SNTA1
), прямо или косвенно влияющих на функцию натриевого канала. Мутации генов
KCNQ1
,
KCNH2
и
SCN5A
, вызывающие LQTS1, 2 и 3 соответственно, являются так называемыми основными генами LQTS, а мутации в них предполагают высокую вероятность врожденного LQTS и имеют важное значение для стратификации риска [24]. Следует отметить различия в частоте встречаемости мутантных вариантов в зависимости от популяции [49].
У пациентов с синдромом
Бругада
(СБ) наблюдается склонность к развитию смертельных аритмий в основном во время сна, при отсутствии ишемии миокарда, нарушений обмена электролитов и иных структурных изменений в сердце. Маркеры ЭКГ при СБ хорошо известны. Семейный анамнез ВСС при СБ довольно типичен. Частота встречаемости СБ в европейской и американской популяциях составляет 1 на 10 000 населения. Установлено порядка 350 различных вариантов мутаций гена, ассоциированных с СБ [50, 51]. Наиболее часто СБ ассоциируют с мутациями гена
SCN5A
. По мнению многих авторов [24, 52, 53], в 65% случаев генетический субстрат при БС не установлен.
Катехоламинергическая полиморфная
желудочковаятахикардия
(КПЖТ) предполагает нарушение внутриклеточного гомеостаза Ca2+. Данный вид КМП связывают с мутациями гена
RyR2
, кодирующего рецепторы рианодина, ответственные за выделение кальция из саркоплазматической сети кардиомиоцитов. Мутация наследуется по аутосомно-доминантному типу. По аутосомно-рецессивному типу наследуются мутации гена
CASQ2
, ответственного за синтез белка кальсеквестрина, выполняющего функцию связывания Ca2+ в саркоплазматической сети. Еще одна мутация, связанная с КПЖТ, известна в гене
TRDN
, кодирующем триадин (белок, связывающийся с кальсеквестрином и RyR2-рецепторами). При наличии мутаций в данных генах ВСС часто наступает во время эмоционального стресса [54] либо физических нагрузок [24].
Проведенный анализ научной литературы позволяет утверждать, что ВСС — одна из основных причин смертности во всем мире. Она связана в основном с ИБС у пожилых людей и различными формами КМП у лиц молодого возраста. Особенность течения болезни у страдающих различными формами КМП объясняется тем, что остро развившаяся сердечная недостаточность часто является первым и единственным симптомом имевшегося заболевания. Секционные исследования умерших молодых людей, а также полученные статистические данные свидетельствуют о том, что в 1/3 случаев при вскрытии или проведении дополнительных гистологических исследований не обнаруживают специфических морфологических маркеров данной патологии. В настоящее время нет точных эпидемиологических данных о частоте встречаемости различных видов КМП в разных популяциях в связи с отсутствием единых строгих критериев диагностики данной патологии [46]. Возможно, что неопределенность в результаты эпидемиологических исследований вносит МКБ-10. Вероятно, в ней должна быть более детально структурирована эпидемиология данной нозологии. В частности, в МКБ-10 отсутствуют разделы, посвященные, например, аритмогенной КМП правого желудочка, что подразумевает использование раздела «Другие кардиомиопатии (I 42.8)» и «Кардиомиопатия неуточненная (I 42.9)». В последнее десятилетие сложилось понимание, что в основе первичных КМП лежат мутации в генах, ответственных за синтез белков, обеспечивающих структуру саркомера, десмосом либо мембранных каналов, обеспечивающих ионный обмен в кардиомиоците. Методы генетического типирования, а именно переход на использование NGS-типирования, позволили расширить представление о природе КМП, особенно в случаях, когда не определен морфологический субстрат [17]].
Авторы статьи не сомневаются, что проведенное после смерти молекулярно-генетическое типирование на предмет носительства мутаций в генах, ассоциированных с ВСС, имеет право на внедрение в практическую работу судебно-медицинского эксперта.
Следует отметить, что при большом количестве установленных мутаций невольно возникает проблема интерпретации полученных результатов генетического типирования в практике судебно-медицинского эксперта. Некоторые авторы [55] объективно высказывают пессимизм в экспертной оценке полученных генетических результатов. По нашему мнению, один из путей, позволяющий объективно интерпретировать полученные генетические данные, — это тщательный комплексный анализ обстоятельств наступления смерти молодого пациента (потерпевшего), собранного семейного анамнеза, данных вскрытия и результатов лабораторного исследования.
Авторы заявляют
оботсутствии конфликта интересов.
1e-mail; https://orcid.org/0000-0001-5370-4931